

计算催化自由能台阶图是电催化研究中的核心方法之一,它通过展示反应路径中各步骤的自由能变化,帮助研究人员理解反应机理、确定决速步骤,并评估催化剂的性能。以下华算科技将从理论基础、计算步骤、关键参数、软件工具及实际应用等方面进行详细分析,并结合多张示意图进行说明。



理论基础
自由能台阶图(Free Energy Diagram)是一种用于描述催化反应路径中各步骤能量变化的图形工具。它通过将反应物、中间体和产物的吉布斯自由能(Gibbs Free Energy, ΔG)以阶梯状形式展示,直观地反映反应路径中的能量起伏。
自由能台阶图不仅能够揭示反应的热力学驱动因素,还能帮助识别反应的决速步骤(Rate-Determining Step, RDS),从而为催化剂设计提供理论依据。
在电催化反应中,自由能台阶图通常用于分析析氢反应(HER)、析氧反应(OER)、氧还原反应(ORR)、二氧化碳还原反应(CO2RR)等。这些反应的自由能变化与催化剂的电子结构、吸附能力、过渡态稳定性等因素密切相关。
例如,在HER中,氢中间体(如H*、H2O、OH等)的吸附能是决定反应速率的关键因素;而在OER中,氧中间体(如OOH、O2)的吸附能和脱附能则决定了反应的过电位。
计算步骤
计算自由能台阶图的流程通常包括以下几个步骤:
确定基元反应步骤
基元反应(Elementary Reaction)是描述反应路径中不可逆的最小单元。在电催化反应中,基元反应通常包括吸附、脱附、电子转移等步骤。例如,在HER中,基元反应可能包括:
H⁺的吸附(H⁺+*→H*)
电子的转移(H*+e⁻→H2)
H2的脱附(H2→*)
在OER中,基元反应可能包括:
O2的吸附(O2+*→ O2*)
O2的分解(O2*→O*+O*)
O的脱附(O→*)
确定基元反应步骤是构建自由能台阶图的基础,它决定了后续计算的准确性。
搭建结构模型
在计算自由能台阶图之前,需要构建催化剂的表面模型。这通常包括:
表面Slab模型:将催化剂表面切割成二维平面,模拟其在溶液中的暴露状态。
吸附模型:确定中间体(如H*、OH*、O等)的吸附位点和构型。例如,在Pt(111)表面上,H可能吸附在台阶位置,而OH*可能吸附在凹陷位置。
过渡态模型:通过过渡态搜索(如Nudged Elastic Band, NEB)确定反应路径中的过渡态。
搭建结构模型时,需要考虑催化剂的晶格常数、表面重构、吸附位点的几何构型等因素。例如,在Fe-N4-C单原子催化剂中,N原子的配位环境对H*的吸附能有显著影响。
DFT计算获取结构能量
密度泛函理论(Density Functional Theory, DFT)是计算自由能台阶图的核心工具。通过DFT计算,可以获取以下信息:
吸附能:中间体在催化剂表面的吸附能(ΔG–ads)。
零点振动能:分子在基态下的振动能量,通常通过频率计算(Frequencies)获得。
熵变:反应过程中系统的熵变(ΔS)。
吉布斯自由能:通过热力学修正(如零点振动能和熵变)计算得到的吉布斯自由能(ΔG–Gibbs)。
DFT计算通常包括以下步骤:
结构优化:通过能量最小化算法(如BFGS)优化催化剂表面和中间体的几何构型。
频率计算:计算分子的振动频率,以确定零点振动能。
能量计算:计算反应物、中间体和产物的总能量(Total Energy, E–total)。
例如,在NiPc-CN催化剂中,通过DFT计算可以得到不同反应路径的自由能变化,从而绘制出自由能台阶图。
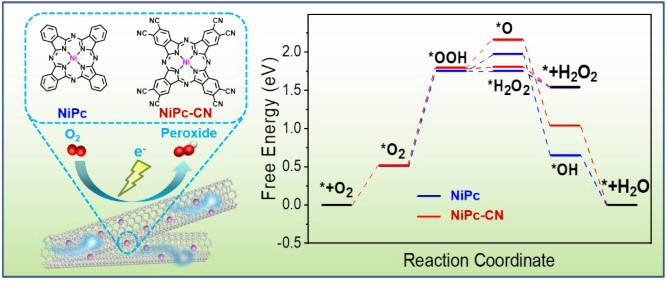
绘制自由能台阶图
在获得各步骤的吉布斯自由能后,可以将初始态的自由能设为0,绘制出自由能台阶图。通常,自由能台阶图的横轴表示反应路径,纵轴表示吉布斯自由能(单位为eV)。每个台阶代表一个反应步骤,其高度表示该步骤的自由能变化。
例如,在Fe-NH MOF催化的ORR反应中,自由能台阶图显示其过电位为0.38V,优于Fe-O和Fe-S体系。这表明Fe-NH MOF在ORR反应中具有更好的催化性能。
关键参数与修正
在计算自由能台阶图时,需要考虑以下关键参数:
零点振动能(Zero-Point Vibrational Energy,ZPE)
零点振动能是分子在基态下的振动能量,通常通过频率计算获得。在电催化反应中,零点振动能对自由能的修正非常重要。例如,在HER反应中,H*的吸附能需要通过ZPE进行修正,以获得更准确的吉布斯自由能。
熵变(Entropy Change,ΔS)
熵变是反应过程中系统的熵变,通常通过热力学计算获得。在电催化反应中,熵变对自由能的修正同样重要。例如,在OER反应中,OOH*的脱附能需要通过熵变进行修正,以获得更准确的吉布斯自由能。
标准氢电极(Standard Hydrogen Electrode,SHE)模型
在计算自由能台阶图时,通常需要使用标准氢电极模型(Norskov模型)来关联电极电位与自由能。例如,在HER反应中,H*的吸附能可以通过SHE模型进行计算,以获得更准确的吉布斯自由能。
温度修正(Temperature Correction)
在实际反应中,温度对自由能的修正非常重要。例如,在HER反应中,H*的吸附能需要通过温度修正进行计算,以获得更准确的吉布斯自由能。
软件工具与计算方法
计算自由能台阶图通常需要使用第一性原理计算软件,如VASP、Materials Studio、Gaussian等。这些软件提供了从结构建模、能量计算到自由能修正的完整工具链。
VASP
VASP是一种广泛使用的第一性原理计算软件,适用于金属、半导体和绝缘体的电子结构计算。在电催化反应中,VASP可用于计算催化剂表面的吸附能、过渡态和自由能。例如,在NiPc-CN催化剂中,通过VASP计算可以得到不同反应路径的自由能变化。
Materials Studio
Materials Studio是一种集成化的分子建模和模拟软件,适用于表面吸附、过渡态搜索和自由能计算。在电催化反应中,Materials Studio可用于构建催化剂表面模型、计算吸附能和自由能。例如,在Fe-NH MOF催化的ORR反应中,通过Materials Studio可以绘制出自由能台阶图。
Gaussian
Gaussian是一种量子化学计算软件,适用于分子和材料的电子结构计算。在电催化反应中,Gaussian可用于计算催化剂表面的吸附能、过渡态和自由能。例如,在Ni-SAC@NFC催化剂中,通过Gaussian计算可以得到CO2还原反应的自由能变化。
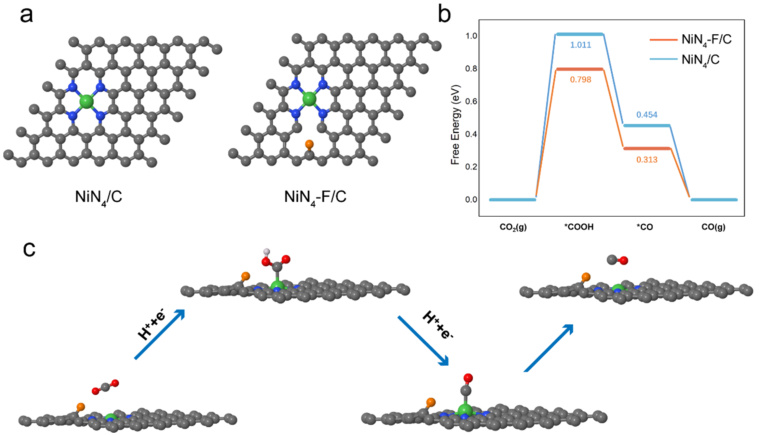
实际应用与案例分析
自由能台阶图在电催化反应中的应用非常广泛,以下是一些典型的应用案例:
析氢反应(HER)
在HER反应中,自由能台阶图用于分析H的吸附能和脱附能。例如,在Pt(111)表面上,H的吸附能可以通过自由能台阶图进行计算,以评估催化剂的活性。在CoN3-TeN1双原子催化剂中,通过优化电子结构,可以降低H脱附能垒,提升反应速率。
析氧反应(OER)
在OER反应中,自由能台阶图用于分析O2的吸附能和脱附能。例如,在IrO2/SrIrO3异质结构中,通过优化吸附能差值,可以显著降低过电位,突破传统理论限制。在Fe-N4-C单原子催化剂中,通过自由能台阶图可以预测产物选择性。