

晶体的能带结构
晶体中大量原子(分子、离子)规则排列成点阵结构,晶体中形成周期性势场。由于晶体中原子的周期性排列,价电子不再为单个原子所有。共有化的电子可以在不同原子中的相似轨道上转移,可以在整个固体中运动。
对于原子的外层电子(高能级),势垒穿透概率较大,属于共有化的电子;原子的内层电子与原子核结合较紧,一般不是共有化电子。

图1. 晶体与非晶体材料结构对比。DOI: 10.3390/electronics4030424
能带的形成
由于晶体中各原子间的相互影响,原来各原子中能量相近的能级将分裂成一系列和原能级接近的新能级,这些新能级基本上连成一片,形成能带(energy band)。
晶体的能带结构常常采用近自由电子模型来描述,能带电子被看作仅受离子实的周期势场的微扰。该模型能够给出关于金属中电子行为的几乎所有定性问题的答案。布洛赫(Bloch)证明了对于含周期性势场的薛定谔方程,其解必定具有如下形式:
Ψk(r)=uk(r)exp(ikr)
式中,uk (r)=uk (r+T),T表示晶格平移矢量。该式表明,对于周期势场中的波动方程而言,其本征函数为一个平面波exp(ik⋅r)乘上一个具有周期性的函数uk(r)。
上述给出的单电子的波函数就是一个所谓的布洛赫函数。布洛赫函数可以叠加为波包,从而表示在离子实势场中自由传播的电子。将Ψk代入薛定谔方程,并取周期性边界条件即可求出Ψk的特征值Ek。
能带结构是Ek-k关系曲线,这种函数空间称为k空间(也称倒易空间、动量空间),它实际上是我们熟悉的空间的Fourier变换。Bloch分子轨道其实就是构成晶格的原子轨道的Fourier变换,函数变量由j变换成k。
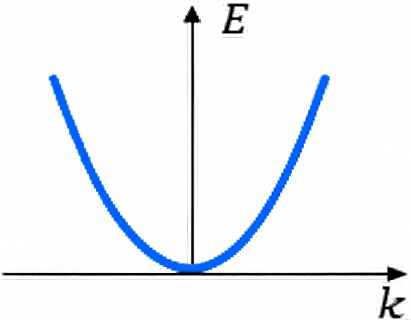
图2. 自由电子的E–k曲线。
当N个原子靠近形成晶体时,由于各原子间的相互作用,原来孤立原子的一个能级,就分裂成N条靠得很近的能级。使原来处于相同能级上的电子,不再有相同的能量,而处于N个很接近的新能级上。
能带宽度ΔE一般为几个eV,能带中相邻能级的能差约为10−22eV。一般而言,外层电子共有化程度高,能带较宽;相应内层电子的能带较窄。
图3. PMN-PT在未极化的8R域状态下,极化后在4R工程化域状态下的倒易空间图像。DOI: 10.1002/adma.202504143
能带中电子的排布
能带中电子的排布服从泡利不相容原理和能量最小原理。N个孤立原子的能级Enl,最多能容纳2N(2l+1)个电子,其中l为角量子数;该能级分裂成N条能级组成的能带后,最多能容纳2N(2l+1)个电子。例如,1s、2s能带,最多容纳2N个电子;而2p、3p能带,最多容纳6N个电子。
根据电子的排布情况,可将能带分为满带、导带、空带和禁带。根据材料的能带结构,可推断材料属于金属、半金属、半导体还是绝缘体,进而推断材料的导电性能。
满带:能带中各能级都被电子填满。满带中的电子由原占据的能级向带内任一能级转移时,必有电子沿相反方向转换,因此,不会产生定向电流,不能起导电作用。
导带:被电子部分填充的能带。在外电场作用下,电子可向带内未被填充的高能级转移,但无相反的电子转换,因而可形成电流。
价带:价电子能级分裂后形成的能带。有的晶体的价带是导带,有的晶体的价带也可能是满带。
空带:所有能级均未被电子填充的能带。由原子的激发态能级分裂而成,正常情况下空着;当有激发因素(热激发、光激发)时,价带中的电子可被激发进入空带;在外电场作用下,这些电子的转移可形成电流。所以,空带也是导带。
禁带:在能带之间的能量间隙区,电子不能填充。
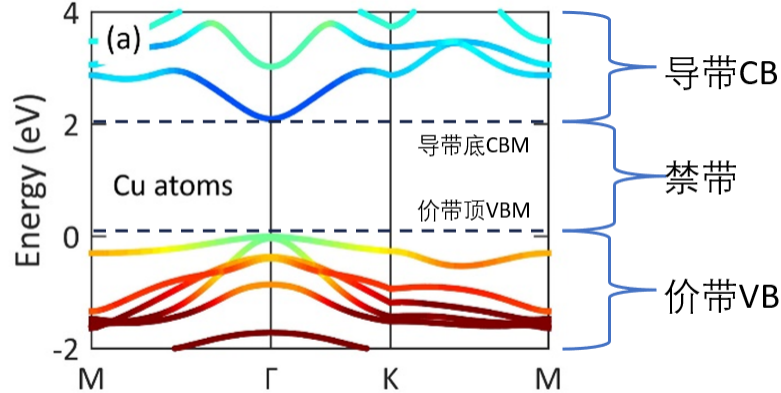
图4. 典型的能带结构示意图。
态密度
能带结构的k空间描述方式在实际应用中常显得不够直观,另一常用的能带结构表示方法是态密度(density of states,DOS)。
对于给定的能带n,其态密度Nn(E)为:
Nn(E)=∫δ[E−En(k)]dk/4π3
式中,En(k)为n能带的色散,态的总密度N(E)是通过在布里渊区内积分求得对所有能带求和得到的。N(E)从负无穷大到积分至费米能级可求得体系中的电子总数。
在自旋极化系统中,可以为自旋向上和自旋向下的电子引入单独的DOS,其和值产生总DOS,它们的差值称为态的自旋密度。DOS(E)代表整个Brillouin区的平均能级密度分布,不具有正负对称性与空间方向性。
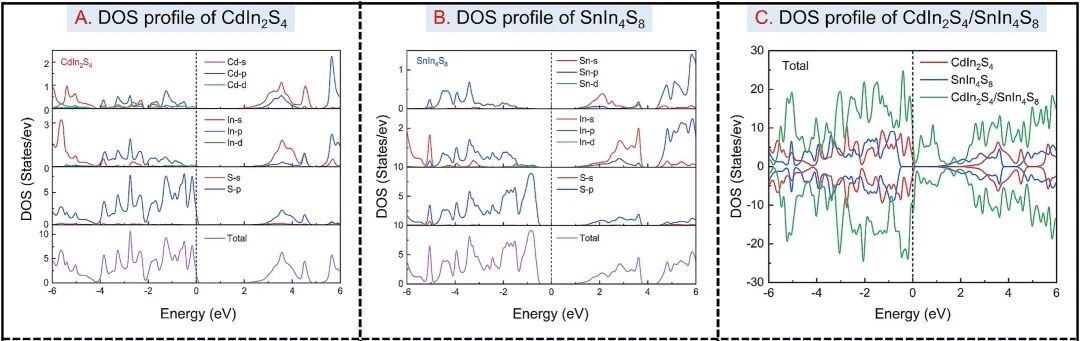
图5. Cdln2S4,Snln4S8,Cdln2S4/Snln4S8的DOS曲线。DOI: 10.1016/j.cej.2025.161413
态密度的另一种表示方式是:Nn(E)dE与能量范围E至(E+dE)内第n个能带中的波矢k成正比,即能带中的能级数目在线性势能标度(E)上的分布,代表能带中的能级数目n随势能E的变化率:
DOS(E)=dn(E)/dE
在Ek-k能带结构中,k值是能级的编号,在dE范围内的k值变化dk应正比于能级数目dn的变化,因此
DOS(E)∝dk/dE
DOS(E)正比于Ek-k曲线的斜率的倒数。换言之,在Ek-k能带结构图中,能带的走向越缓,该Ek下的DOS越大;相反,能带走向越陡,该Ek下的DOS越小。与Ek-k曲线相比,DOS(E)曲线虽然失去了空间细节,但较简单地反映了材料整体的电子结构特征。
DOS(E)是n(E)的微分,因此从带底至EF对DOS(E)曲线进行积分可得到该能带中的电子占有能级数Nf:
Nf=∫EfDOS(E)dE
Nf乘以2便是能带中的填充电子数。
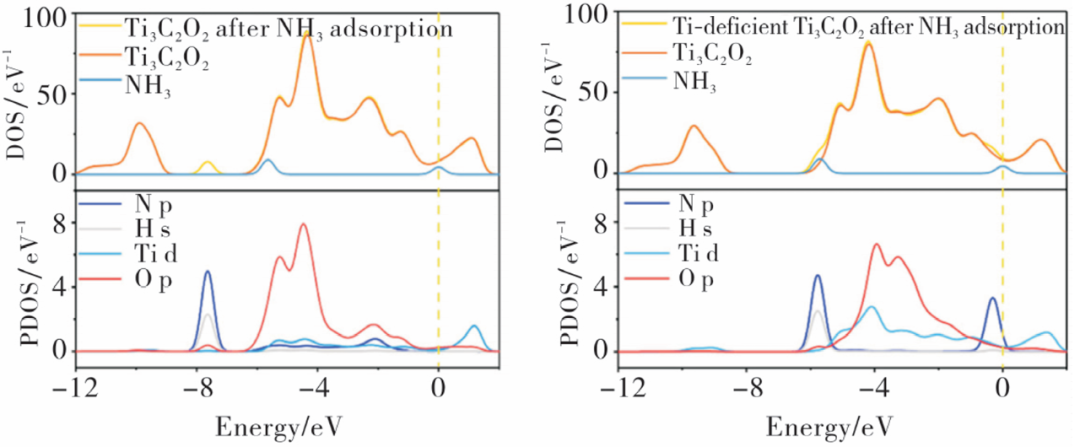
图6. NH3吸附在Ti3C2O2上,NH3吸附在Ti缺陷的Ti3C2O2上的DOS和PDOS。DOI: 10.19411/j.1007-824x.2025.01.001
布居数分析
设分子轨道φ=∑μciμχμ,其中χμ为基矢波函数。定义密度矩阵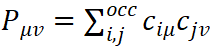
,则有总电子数N=Tr(PS)= ∑μ(PS) μμ,其中Sμν=(χμ∣χν)为重叠积分。电荷qA=ZA−∑μ∈A(PS)μμ。Mulliken布居的优点是简单,可用于定性分析,存在的问题是强烈依赖于基组,对大基组电荷布居偏大。
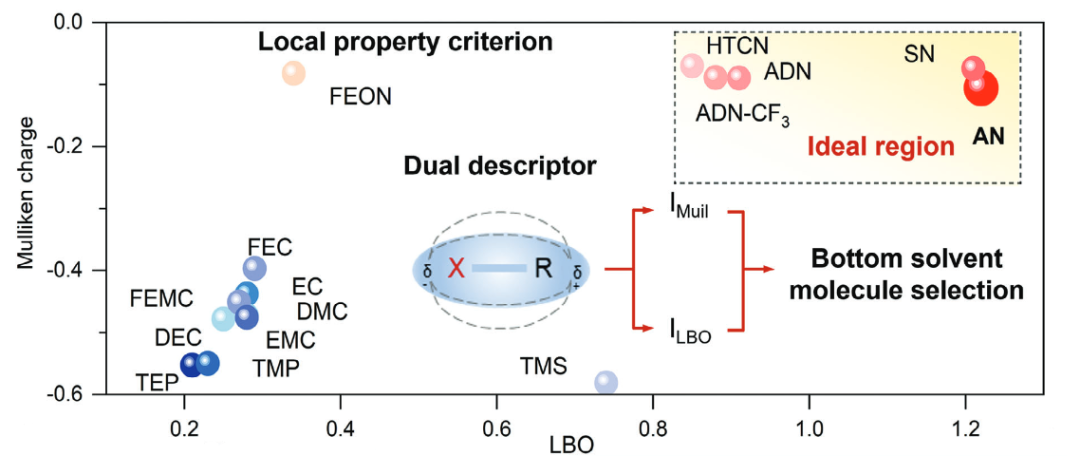
图7. 二维分子选择图,横轴为LBO,纵轴为Mulliken电荷。DOI: 10.1002/adma.202417076
Löwdin布居
相对Mulliken布居进行了小的改进,先对基组作正交化:χr=∑SSrs1/2ϕs——对基矢的依赖变小。
静电势拟合(ESP fitting)
为了正确地描述分子与其他分子的静电相互作用,电荷布居应该重新产生出在分子周围一定区域内的静电势:
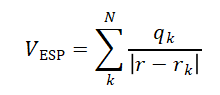
存在的问题:(1)柔性分子的电荷布居具有构象依赖性;(2)内部原子与外势不匹配。
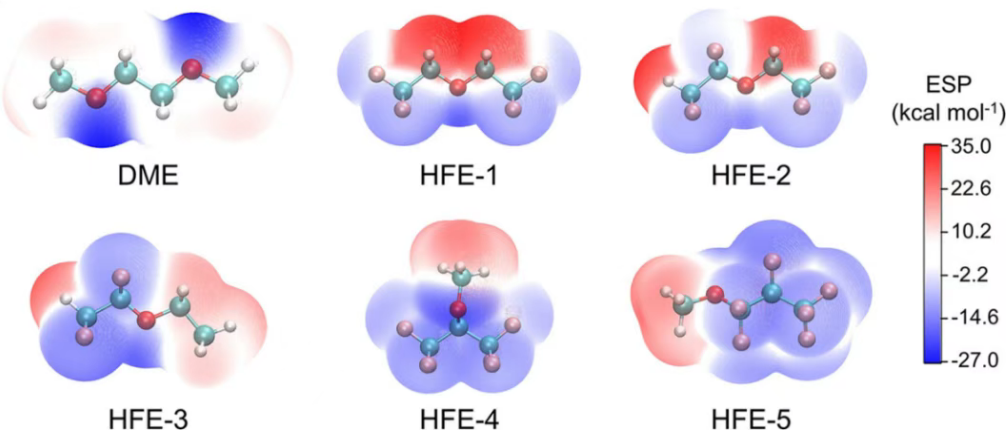
图8. DME和五种氢氟醚(HFEs)的分子静电势。DOI: 10.1021/jacs.3c12358
Hirshfeld布居
ρd=ρ(r)−∑αρα(r−Rα)
其中等式右边第一项为形成分子时的电荷密度,第二项为单个原子的电荷密度叠加。电荷qα=∫ρd(r)Wα(r)dr,其中权重Wα(r)=ρα(r−Rα)[∑βρβ(r−Rβ)]−1。
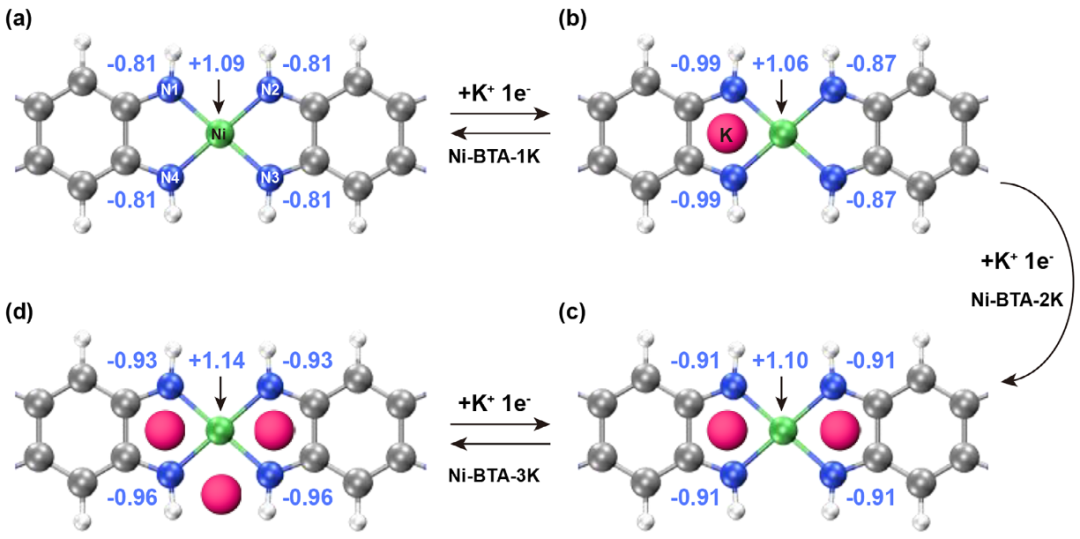
图9. Hirshfeld电荷分析定量地证明了氧化还原活性主要源于配体,而Ni中心起到了稳定结构和缓冲电荷的作用。DOI: 10.1002/adma.202509022
根据电荷分布的拓扑结构进行分析,梯度为零的地方就是分界面。
∇ρ⋅n=0
总的来说,所有的布居分析都是定性的。
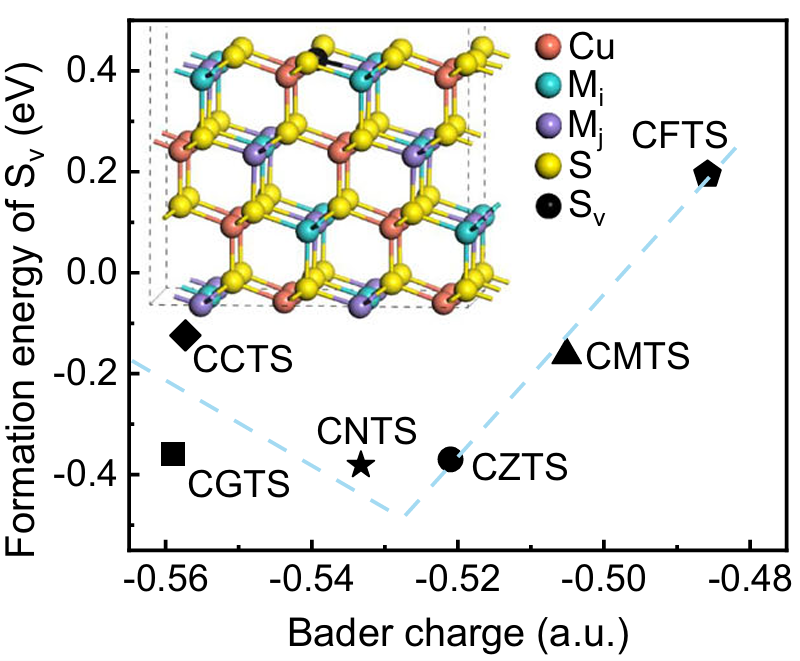
图10. 在Cu2MiMjS4薄膜中,随着Cu原子的Bader电荷变化,硫空位(Sv)的形成能图,呈现倒火山关系。DOI: 10.1038/s41467-025-61850-7
其他性质
电荷密度和差分电荷密度通常可用来直接判断成键性质及电子盈亏,以及共价键、离子键、金属键、氢键、分子间互相作用。电离能(IP)为拿走一个电子所需要的能量,电子亲和能(BA)为得到一个电子所释放的能量。对金属,可以定义功函数为电子从金属逃逸时所需要克服的功。在计算中,真空能级等于Veff→∞时的值,为了加快收敛,通常只考虑静电部分。