

π-π相互作用是芳香体系间通过π电子云离域产生的非共价作用力,其本质是量子力学框架下的多电子效应,主要表现为静电、色散与轨道耦合的协同作用。
在材料科学、生物识别与催化领域具有核心地位,如MOF材料中的离子传输强化与聚合物光催化剂的电荷分离调控。
密度泛函理论(DFT)计算揭示其能量范围在-0.3至-0.5 eV(弱于氢键但强于范德华力),且几何构型(堆叠距离与取向)是决定作用强度的关键参数。本文从物理本质、计算方法和功能应用三层次系统解析其微观机制。
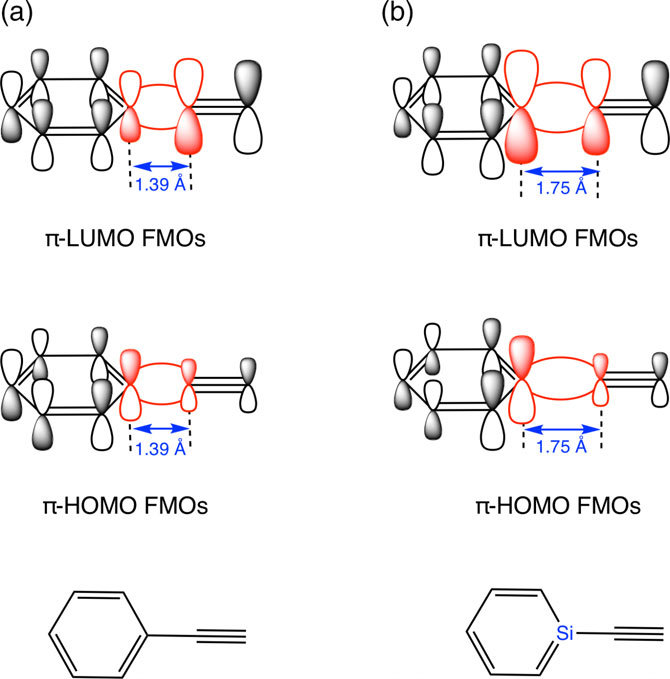

π-π相互作用的物理本质
π-π相互作用的量子力学基础源于离域π电子体系的四极矩特性。芳香环(如苯)虽无净偶极矩,但因对称性形成电四极子:环平面带正电,环上方与下方π电子云形成负电区域。
当两个芳香体系靠近时,一个环的负电区域与另一环的正电平面产生静电吸引,同时伴随π电子云的轨道重叠。这种耦合作用显著降低体系总能量,例如苯二聚体的结合能约为-8 kJ/mol,而扩展共轭体系(如芘)可提升至-30 kJ/mol以上。
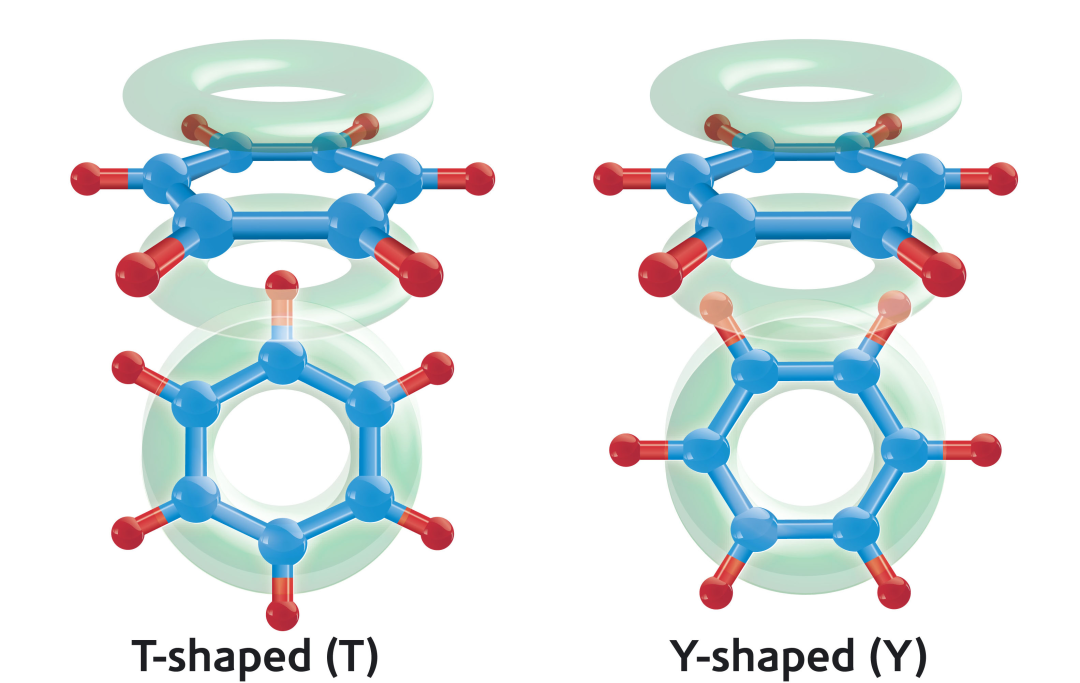
空间构型与电子密度的协同作用是调控π-π强度的核心变量。平行堆叠构型(如石墨烯层间)以色散力主导,距离通常为3.3-3.8 Å,能量随共轭面积增大而增强;T型构型(如蛋白质中苯丙氨酸侧链)则依赖静电互补,其中一个芳环的C-H键(δ+)垂直指向另一芳环的电子富集区(δ-)。
此外,取代基效应通过改变电子密度分布显著调谐作用力:吸电子基(-NO₂)削弱堆叠作用,供电子基(-OCH₃)则增强π电子供给能力。
DOI: 10.1021/acs.cgd.8b01630
阳离子-π作用作为π-π作用的特殊衍生形式,体现了电荷–四极矩耦合机制。当阳离子(如Na⁺、K⁺)靠近芳环时,其正电荷与π电子云的负电区域产生强静电吸引,结合能可达-80 kJ/mol(如Na⁺-苯体系),接近氢键强度的两倍。
该作用在酶催化活性中心(如乙酰胆碱受体结合位点)和离子筛分膜(如MOF中的选择性离子传输通道)中起关键定向作用。
理论就按揭示π-π作用机制
密度泛函理论(DFT)是解析π-π相互作用的基石工具,其核心优势在于平衡精度与计算效率。通过选择适当泛函(如M06-L、ωB97XD)可准确描述长程色散校正,突破传统泛函(如PBE)对弱作用力低估的局限。
例如联苯烯(BpN)吸附三氯乙烯(PCE)的DFT计算中,采用D3色散校正后吸附能从-0.22 eV提升至-0.48 eV,与实验值偏差。
DFT还可量化电子结构响应:如Mn-MOF的π堆叠通道使Zn²⁺扩散能垒降至0.073 eV(SO₄²⁻为1.50 eV),揭示离子选择性传输的电子本质。
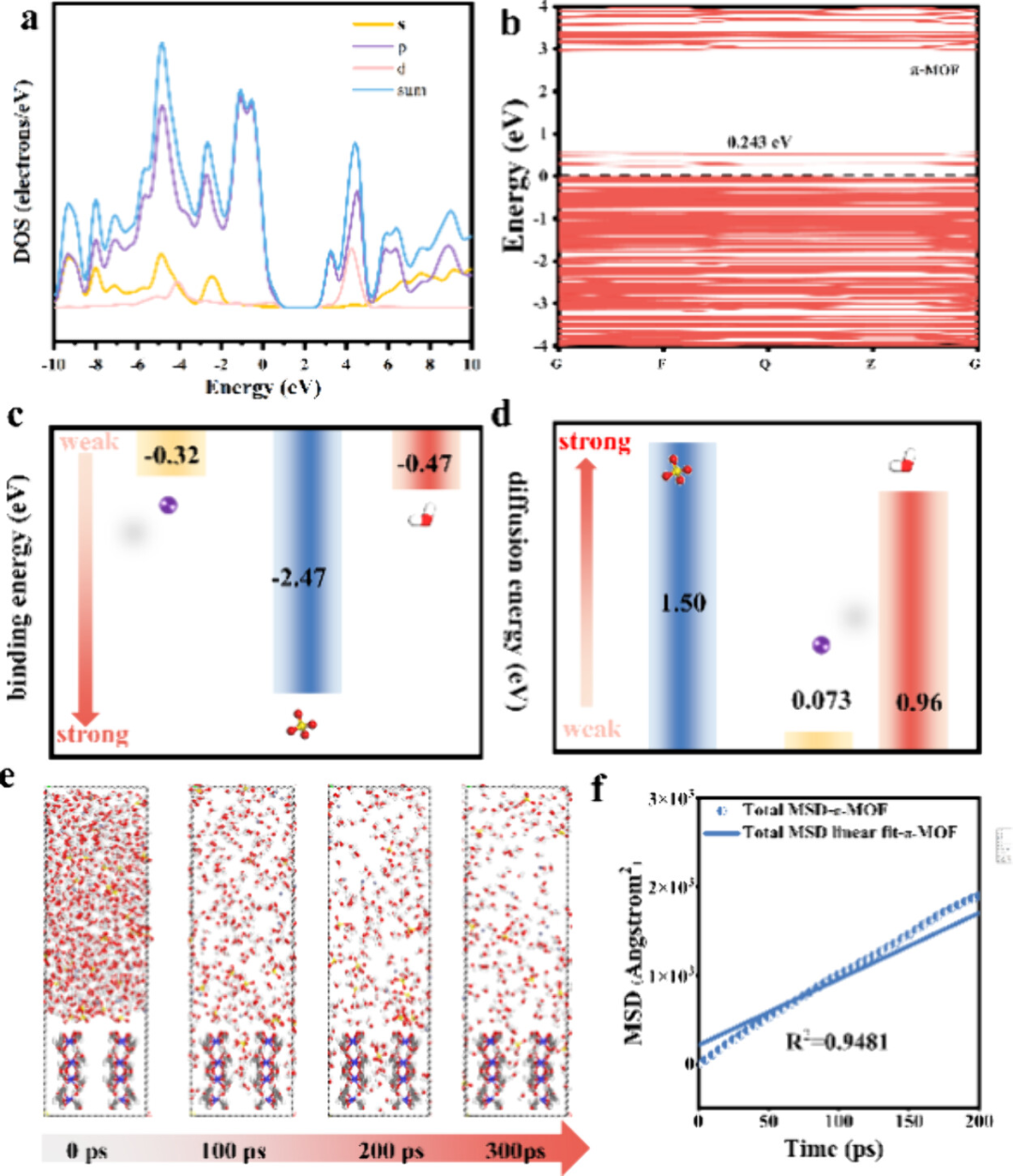
DOI:10.1021/acs.jpclett.4c01780
前沿轨道与电子密度分析是解构作用机制的直接手段。通过投影态密度(PDOS)与晶体轨道哈密顿布居(COHP)分析,可可视化轨道耦合效应。
例如双核Cu(I)配合物[Cu₂(bpe)(bipy)₂(PCHO)₂]²⁺的DFT计算显示:HOMO由Cu的d轨道与配体π轨道杂化形成,LUMO则定位于bpe配体的反键π轨道,能隙1.64 eV;π-π堆叠使HOMO电子密度向相邻芳环离域,降低重组能并增强荧光量子产率。
此外,电子密度差分图可直观显示π-π界面处的电荷转移(通常),如聚合物氮化碳(PCN)层间存在电荷转移通道,诱导HOMO与LUMO空间分离(VBM与CBM分属不同七嗪单元)。
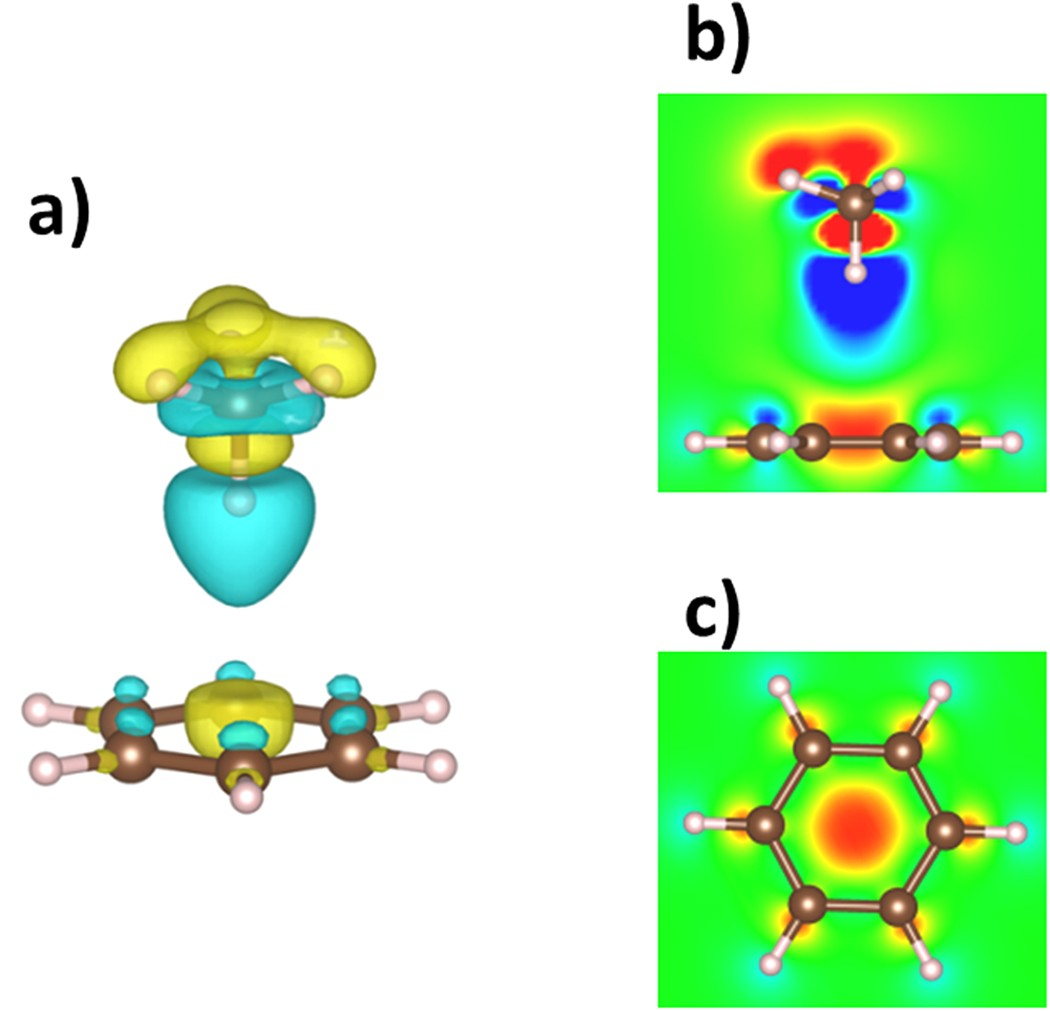
DOI:10.1038/srep22304
激发态与非绝热动力学的拓展模拟深化动态过程认知。含时密度泛函理论(TD-DFT)可预测π-π体系的光响应特性:如噻吩–钠离子夹心复合物(Na⁺-2T)在380 nm处出现MLCT吸收峰,对应S₀→S₁跃迁(振子强度f=0.15),证明阳离子-π作用可调控光学带隙。
非绝热动力学(NAMD)则揭示PCN中载流子寿命:层间π-π作用使电子弛豫加速至飞秒级(较单层快3倍),因堆叠结构提供额外的声子散射通道。
量子化学弱相互作用分析方法:量子化学的能量分解分析(EDA)和非共价相互作用指数(NCI)为解构π-π作用提供了原子级分辨的物理图景。
在EDA框架下(如SAPT、ALMO-EDA),苯二聚体的总结合能(-8.2 kJ/mol)可分解为色散力(-18.3 kJ/mol)、静电作用(-4.1 kJ/mol)、泡利排斥(+14.2 kJ/mol)及轨道极化项(-0.5 kJ/mol),明确揭示色散力的主导地位。
而NCI方法通过电子密度梯度∇²ρ的可视化,直接呈现π-π界面处的弱吸引区域(∇²ρ且λ₂),如石墨烯层间距3.4 Å处出现的盘状绿色等值面,对应色散作用的空间分布。
对于取代芳环体系(如硝基苯–苯酚复合物),EDA显示吸电子基使静电吸引力降低35%,而NCI图谱中π-π接触面显著收缩,与结合能减弱形成定量关联。