

分子描述符是将复杂的化学信息转化为可量化数据的数值表示,从而实现分子性质的数学处理。它们在化学、药物科学和材料设计等领域发挥着基础性作用。量子化学描述符是分子描述符的一个重要类别,它们源自量子力学计算,能够深入揭示分子的电子结构、反应活性以及其他通过简单经验方法难以获得的内在分子性质。

分子反应活性与量子化学描述符
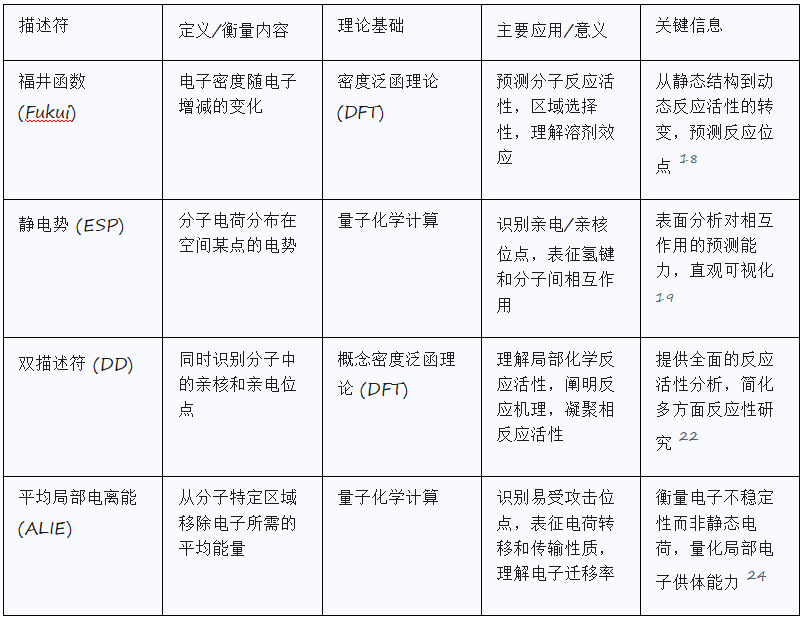
反应活性描述符,通常源自密度泛函理论(DFT),旨在识别分子中易发生化学攻击(亲电或亲核)或电子转移的位点。
福井函数:揭示亲电和亲核位点
福井函数描述了电子密度在增加或移除一定量电子后的变化。它是密度泛函理论中的一个基本概念,广泛用于预测分子的反应活性。通过分析福井函数,研究人员可以识别最活泼的位点。
亲电攻击 (f−(r)): 表示移除电子后电子密度的变化 (f−(r)=ρN(r)−ρN−1(r))。这表明了易受亲核攻击的位点。
亲核攻击 (f+(r)): 表示增加电子后电子密度的变化 (f+(r)=ρN+1(r)−ρN(r))。这表明了易受亲电攻击的位点。
福井函数在多个方面具有应用价值:
预测区域选择性: 对于理解反应在分子中优先发生的位点至关重要,从而影响产物的收率和性质。例如,在亲电芳香取代反应(如甲苯硝化)和过渡金属络合物的反应活性研究中,福井函数能够准确预测反应位点。
理解溶剂效应: 通过计算不同溶剂环境中的福井函数,研究人员可以深入了解溶剂如何调节溶质的电子密度和反应活性。
福井函数超越了传统描述符对静态分子性质的描述,引入了动态视角,通过量化电子转移时电子密度的“变化”来直接联系化学反应活性。这种动态观点对于预测反应机理和结果至关重要,它使研究人员能够预测分子中特定区域在化学反应中如何表现,而不仅仅是识别“富电子”或“贫电子”区域。这提供了对化学选择性更细致的理解。
静电势(ESP):绘制分子相互作用和反应活性图
静电势(ESP)是衡量分子电荷分布在空间中某一点产生的电势的量度。它最容易通过量子化学方法对具有明确电子密度的单个分子进行计算。
ESP对于理解化学反应活性、原子结构和分子间相互作用至关重要。其应用包括:
识别亲电/亲核位点: 负ESP区域表示亲核位点(富电子,易受亲电攻击),而正 ESP表示亲电位点(贫电子,易受亲核攻击)。
表征氢键和分子间相互作用: ESP图用于分析非共价相互作用。
推导性质: 原子和阴离子半径、电负性和能量可以从ESP中推导出来。
可视化电荷分布: ESP图可以显示在电子密度表面上,提供电荷分布的指示。
ESP分析的一种重要方法是考虑分子表面上的ESP值。这突出了尽管ESP是一个三维场,但其在分子表面上的值特别具有信息量。表面是分子间相互作用的第一个接触点。这强调了表面性质在分子识别、结合和溶剂相互作用中的重要性。它允许对一个分子如何“看待”另一个接近的分子进行直观的理解和可视化,从而指导药物发现等领域的合理设计。
双描述符(DD):同时识别反应中心
双描述符(DD),是一种反应活性指数,能够同时识别分子系统中的亲核和亲电位点 。其计算明确涉及从参考系统中添加和移除一个过量电子,使其特别适用于理解与电子转移相关的反应活性。
DD的应用包括:
局部化学反应活性: 通过突出亲核性和亲电性来理解局部化学反应活性。
阐明反应机理: 可用于阐明有机化学反应机理。
凝聚相反应活性: 已证明在研究DFT模拟液态水与水合电子的反应活性方面具有实用性,能够预测哪些特定的水分子将参与反应 。
与通常为亲电或亲核攻击单独计算的福井函数不同,双描述符能够“同时”识别这两种类型的反应位点 。这从单次计算中提供了分子潜在反应路径的更全面和整合的视图。这种效率和全面性使得DD成为初步筛选或机制研究的强大工具,尤其是在两种反应活性都相关的情况下。它简化了分析过程,并能揭示分子内不同反应中心之间微妙的相互作用。

DOI:10.1021/acs.jctc.4c00580
平均局部电离能(ALIE):表征电子不稳定性
平均局部电离能(ALIE),I(r),表示从分子内特定区域移除一个电子所需的平均能量。其最低值揭示了电子束缚最不紧密的区域,从而指示了与亲电试剂或自由基反应的有利位点。它还能在一定程度上表征电荷转移行为和传输性质。
ALIE 的应用包括:
反应行为: 识别电子束缚较弱、因此更具反应活性的区域(电子供体)。
基础领域: 在理解原子壳层结构、电负性、局部极化率和硬度方面发挥重要作用。
电子迁移率: 受体中局部均匀的ALIE分布可以带来更高的电子迁移率,这对于材料设计至关重要。
与 MEP 互补: 常与分子静电势(MEP)结合使用,以评估易受亲电或亲核攻击的分子区域。
与静电势(ESP)提供电荷分布的静态图像不同,ALIE 提供了从特定区域移除电子的“难易程度”信息。这是一种更具动态性和能量的电子可用性度量,直接与电离过程和电子转移相关联。低 ALIE 值意味着电子“束缚较弱”,使其更具反应活性。这种区别对于理解氧化还原反应、自由基化学以及设计以电子传输为关键的材料(例如,有机半导体、光伏电池)至关重要。它提供了局部电子“供体能力”的更直接衡量。
DOI:10.1021/acsomega.4c10273
量子化学反应活性描述符总结
电子结构描述符
电子结构描述符表征分子内电子的分布和能量,直接影响其稳定性、光学性质和相互作用。
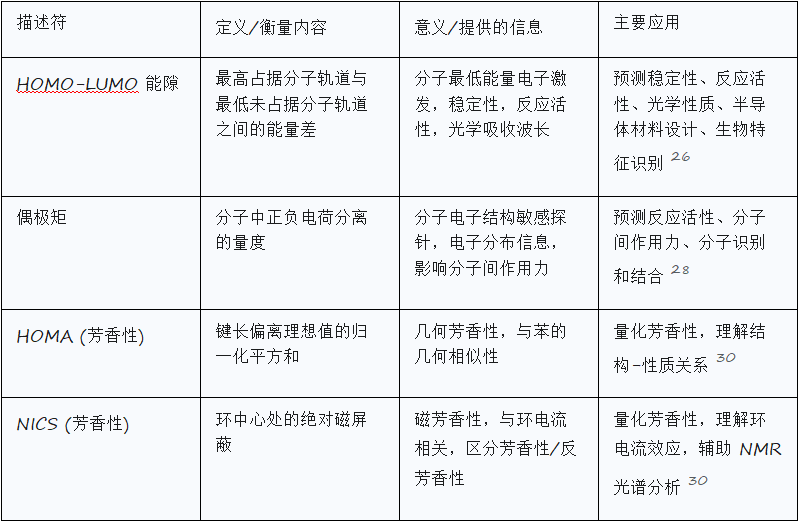
HOMO-LUMO能隙:稳定性、反应活性和光学性质
HOMO(最高占据分子轨道)和LUMO(最低未占据分子轨道)是前沿轨道。HOMO-LUMO能隙是它们之间的能量差。通常,HOMO-LUMO能隙代表分子中可能发生的最低能量电子激发。
HOMO-LUMO能隙的应用包括:
稳定性与反应活性: 较小的HOMO-LUMO能隙通常表示化合物稳定性较低、反应活性较高。它可用于预测结合亲和力。
光学性质: HOMO-LUMO能隙的大小可以指示化合物能够吸收哪些波长的光,从而影响其颜色。
半导体: 在有机半导体中,HOMO能级类似于价带最大值,LUMO能级类似于导带最小值。
有机金属化学: LUMO瓣的大小有助于预测π配体上的加成位点。
生物特征: HOMO-LUMO能隙与亲水性结合,可作为区分生物和非生物化学的生物特征。
HOMO-LUMO能隙是一个纯粹的量子力学概念,源于分子轨道理论。然而,它直接预测了宏观性质,如颜色(光学吸收)和稳定性,甚至对生物特征识别也有影响。这体现了基本量子描述符如何连接微观和宏观领域。因此,该描述符在材料科学中设计新型有机电子产品(半导体)以及在天体生物学中检测生命迹象方面具有不可估量的价值。它与电子激发的直接联系使其成为理解光化学和光谱学的基石。

偶极矩:量化电荷分离和分子间作用力
偶极矩是衡量分子中正负电荷分离程度的量度,是一个具有大小和方向的矢量。它在量子力学中定义为偶极算符对分子波函数的期望值。
偶极矩是分子电子结构的敏感探测器,提供有关分子内电子分布的宝贵信息。计算偶极矩的准确性取决于波函数的质量。其应用包括:
反应活性和分子间相互作用: 大偶极矩表示显著的电荷分离,这会影响分子对其他物种的反应活性。
分子间作用力: 偶极矩是决定偶极-偶极和偶极-诱导偶极相互作用强度和性质的关键因素。
分子识别和结合: 通过调节分子间的相互作用力,偶极矩影响生物系统中的分子识别和结合亲和力。
偶极矩量化了电荷分离,这种分离直接决定了分子的极性,进而控制其与其它分子进行静电相互作用的能力。相互作用能量的方程明确显示了对偶极矩的平方依赖性,表明其具有强大影响力。这使得偶极矩成为预测在不同溶剂中的溶解度、膜渗透性以及药物-受体结合的关键描述符。它是衡量分子在化学环境中“社会行为”的简单而强大的指标。
芳香性分析:HOMA和NICS指数用于环状离域
芳香性是一个无法直接观测但至关重要的化学概念,通过各种指数(几何、磁性、能量、电子、拓扑)来描述。它对于理解反应活性和稳定性至关重要。
HOMA(谐振子芳香性模型):
定义: 一种几何芳香性指数,定义为键长与理想值平方偏差的归一化。芳香族化合物的 HOMA 值为 1,非芳香族为 0。
基础: 简单,基于可观测的键长(实验或计算)。表达与苯的几何相似性。
扩展: 可基于电子或磁性性质(例如,键临界点处的电子密度、化学位移)来提供与苯的电子或磁性相似性。
NICS(核无关化学位移):
定义: 一种计算方法,计算环中心处的绝对磁屏蔽。报告的值符号相反(负值表示芳香性,正值表示反芳香性)。
基础: 与当磁场垂直于环平面施加时,芳香环离域π电子中感应的环电流相关。
应用: 有助于区分芳香族、非芳香族和反芳香族化合物,并评估多环化合物的芳香性。
争议: 在没有原子的点上使用适用于原子的性质来确定,用不可观测的量来衡量不可观测的芳香性。
芳香性被明确指出是“不可观测”且“难以确定”的。然而,它对稳定性和反应活性的重要性使得对其进行量化成为必要。HOMA和NICS从不同角度处理这个问题:HOMA从几何偏差(键长)入手,NICS从磁响应(环电流)入手。这表明需要多方面描述符来表征复杂、抽象的化学现象。
多种芳香性指数的存在,每种都有其优点和缺点(例如,HOMA的简单性与NICS 的磁性基础),表明没有单一描述符能完全捕捉复杂性质的所有方面。研究人员必须根据他们希望研究的芳香性特定方面选择最合适的描述符。对NICS“不可观测”性质的争议也强调了定义和测量理论概念的哲学挑战。
DOI:10.1021/acsomega.4c11451
关键电子结构描述符
电荷分布分析描述符
电荷分布分析描述符提供了分子内电子如何分布的见解,这对于理解键合、反应活性和分子间相互作用至关重要。
原子电荷:比较分析 (NPA, ADCH, Hirshfeld, Mulliken, MK)
原子部分电荷是量子化学中最常用的解释工具之一,代表了描述电荷分布的最简单模型。原子电荷是不可观测的,其定义并非唯一;存在数十种不同的“布居分析”方法。它们最好被视为代理,而非“普遍离子性”的直接测量。
常用方法包括:
Mulliken 布居分析: 最早的方法之一(1955年),但存在固有缺陷,如交叉项的不合理划分和基组依赖性。
Hirshfeld 布居分析: 这种方法,在其原始形式中,通常导致“过小的Hirshfeld电荷”和“分子偶极矩重现性差”。它存在任意性。
原子偶极矩校正的 Hirshfeld (ADCH): 对原始 Hirshfeld的改进,将分子偶极矩的守恒作为约束。ADCH电荷通常大于Hirshfeld电荷,对基组和理论水平不那么敏感,并表现出良好的静电势重现性和与ESP导出电荷的高度相关性。
NPA(自然布居分析)和 MK(Merz-Kollman)电荷: 作为与ADCH比较的常用布居分析方法被提及。
原子电荷的应用包括直观理解基本性质、可能的反应方向以及分子中原子的状态。它们在分子力场中用于静电相互作用计算,并作为分子描述符。
原子电荷“不可观测且定义不唯一”。不同的布居分析(Mulliken、Hirshfeld、NPA、ADCH)对同一分子中同一原子产生不同的数值。这种非唯一性意味着,尽管原子电荷在直观理解电荷分布方面具有吸引力,但其定量值应谨慎解释。
跨不同方法或研究进行比较时,必须考虑所使用的具体布居分析方法。这也推动了ADCH等改进方法的发展,旨在提高重现性并与偶极矩等可观测性质更好地关联。
电子定域函数(ELF)
电子定域函数(ELF)是衡量同自旋电子之间泡利排斥的定量指标。它指示了在另一个同自旋电子附近找到电子的概率。ELF是表征键合的有效工具,通过提供电子分布的生动描述,帮助理解路易斯结构中的电子对定域。
ELF值介于0和1之间:
接近1:共价键形成或孤对电子存在的可能性高(高度定域的电子)。
约0.5:存在金属自由电子。
接近0:电子离域区域。
与原子电荷提供电荷分布的数值不同,ELF提供了电子定域的“空间可视化”表示。这使得化学家能够以数字无法传达的方式“看到”键合模式、孤对电子和离域区域,并直接与直观的路易斯结构概念相关联。
这种可视化特性使ELF成为化学直觉和交流的强大工具。它通过清晰地展示电子定域或离域的位置,有助于理解机理,这对于预测反应活性和设计新分子至关重要。
定域轨道函数(LOL)
定域轨道函数(LOL)是一种基于动能密度的化学键合描述符。它描述了定域轨道重叠以及与电子密度相关的分子键合。LOL表示由轨道梯度产生的最大定域轨道的重叠。当电子定域性高于电子密度时,LOL在相应区域的值会显著高于0.5。
ELF和LOL都是可视化电子定域的工具,但它们基于不同的物理量(泡利排斥与动能密度)。明确指出“ELF阐明了电子对的空间定域,而LOL表示最大定域轨道的重叠。”这表明它们对同一现象提供了互补的视角。
同时使用ELF和LOL可以对化学键合提供更稳健和全面的理解。当其中一个不够清晰时,另一个可以提供额外的见解,从而更自信地解释复杂的键合情况,特别是在挑战性系统如过渡金属络合物或超价分子中。
