范德华校正(Van der Waals correction)在第一性原理计算中具有至关重要的作用,尤其是在处理涉及弱相互作用的体系时。它通过引入范德华力(即色散力、诱导力和取向力)来修正标准密度泛函理论(DFT)中对分子间相互作用的低估问题。以下华算科技介绍范德华校正的详细用途及其在多个领域的应用。
范德华力是一种分子间作用力,主要由瞬时偶极诱导产生的弱引力作用构成。这种力广泛存在于各种体系中,例如惰性气体原子之间、分子晶体中分子之间,以及吸附在固体表面的分子与材料之间。在标准DFT计算中,由于使用的是局部密度近似(LDA)或广义梯度近似(GGA)等泛函,这些泛函通常无法准确描述长程的非局域相互作用,导致对弱相互作用体系的结合能预测出现较大误差。
范德华校正可以通过多种方法实现,主要包括以下几种:
通过预先设定的原子对参数,向DFT能量中加入类似Lennard-Jones形式的–C6/r6色散项(有时加上阻尼函数避免近距离发散)。Grimme等人提出的D2、D3、D3(BJ)以及最新的D4校正等方法,通过引入原子对间的色散系数和截断半径,显著提高了对弱相互作用的描述能力。
构造包含非局域相关项的交换-相关泛函,使其天然地描述色散力。典型例子包括Dion等人提出的vdW-DF泛函及其改进版(如optPBE-vdW、optB88-vdW、rvv10等)。这些泛函在计算过程中直接考虑电子密度之间的长程关联效应,从而自洽产生范德华相互作用。
如Tkatchenko-Scheffler(TS)方法,根据局域环境修正–C6系数,或更高级的方法如DFT+RPA(随机相位近似)和DFT-SAPT(对称适应微扰),但这些计算代价较高。
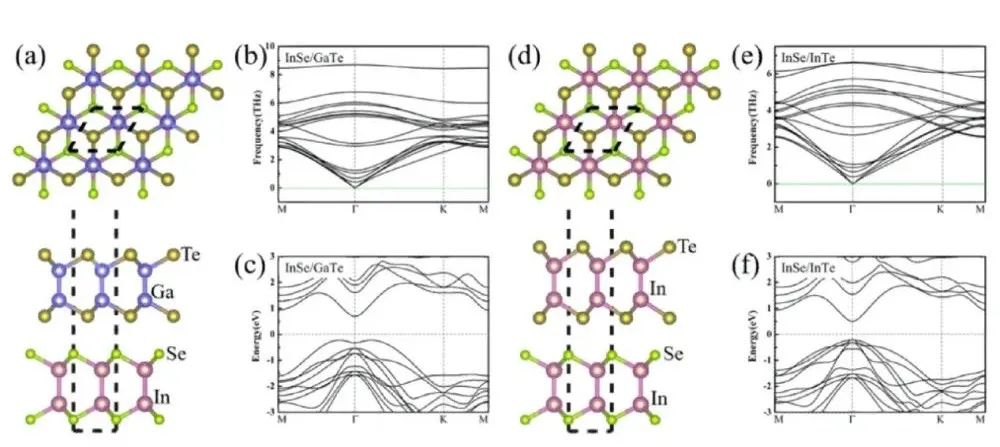
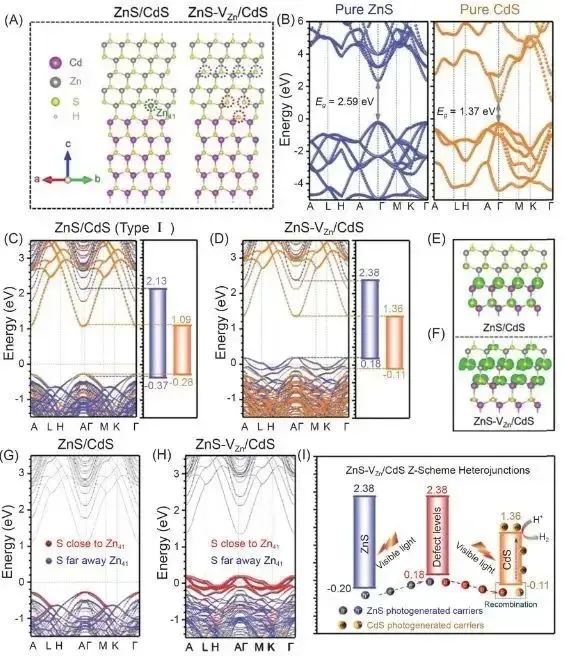
在分子晶体、层状材料(如石墨、MoS2)、吸附体系等,分子或层之间的结合主要由范德华力维系。未考虑色散作用的DFT常常低估这些体系的结合能,导致计算得到的结构过于松散、层间距过大,甚至预测为不相互绑定的状态。例如,对于石墨烯层间相互作用,若不考虑范德华力,两层石墨烯几乎不会有束缚力。
很多化学过程的能量变化很小,范德华相互作用能占有相当比例。如果不加校正,这些能量差的预测将不准确。特别是在分子吸附或化学反应过渡态中,范德华作用可能决定哪一个构型略微稳定。这对于计算吸附能和反应能垒的准确性非常重要。
包含范德华校正的计算通常更接近实验测量。例如,一项研究比较了多种常用DFT泛函对金属表面吸附能的预测,发现引入色散力的泛函(如BEEF-vdW)整体上误差较小,而不含色散的GGA泛函(如PBE)往往低估分子在催化剂表面的吸附强度。
范德华力不仅影响能量,也影响几何结构。没有色散力,计算得到的分子间距离常常偏大,因为少了那股把分子轻轻拉在一起的引力。加入范德华校正后,常常会看到分子更靠近一些,结构参数更符合实际。例如,下文将提到的实例表明,考虑范德华作用后吸附分子的平衡距离显著缩短。
在催化反应中,范德华校正对反应路径和活化能的预测至关重要。例如,在环己烯在Pd表面加氢的反应中,不同范德华校正方法(如DFT-D3、DFT-D3(BJ)、vdW-DF系列泛函、以及rVV10等)对计算的反应路径和活化能的影响显著。研究发现,当范德华作用处理较充分时,反应物和中间体更稳定,导致某一步骤的相对能垒变高;反之亦然。结论是,在异相催化反应的DFT模拟中,范德华校正方法的选择会显著影响定量的活化能预测,从而可能改变对速率控制步骤的判断。
在电化学储能材料研究中,范德华校正对吸附能和离子扩散性质的预测至关重要。例如,在Li2B12H12固体电解质材料的研究中,Malmsev等人使用基于范德华校正的DFT计算,发现温度诱导的[B12H12]2-基团重定向运动主导着材料的有序-无序相变。此外,在Pt低指数晶面上的乙醇吸附研究中,范德华校正显著提高了吸附能的预测准确性,揭示了Pt(110)晶面对乙醇的吸附作用最强。
在二维材料吸附研究中,范德华校正对吸附能的预测至关重要。例如,在MoS2表面的分子吸附研究中,不同范德华校正方法(如D3、D3(BJ)、vdW-DF等)对吸附能的预测结果与实验值最为接近。研究发现,纯PBE显著低估所有分子的吸附能,而加入D3校正和采用vdW-DF泛函的结果与实验值最为接近。
范德华校正是第一性原理计算中不可或缺的一部分,尤其在处理涉及弱相互作用的体系时。通过引入范德华力,可以显著提高计算的准确性,使理论预测更接近实验结果。无论是催化反应、材料吸附、二维材料还是仿生材料,范德华校正都发挥着关键作用。随着计算方法的不断进步,范德华校正将在更多领域展现出更大的应用潜力。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!