

电子结构计算是材料科学、化学、物理等领域中一项基础且重要的研究工具,它通过理论模型和计算方法来预测和分析物质的电子分布、能带结构、态密度、电荷密度等性质。
这些计算不仅有助于理解材料的物理化学性质,还为新材料的设计和开发提供了理论支持。以下华算科技将从电子结构计算的基本原理、常用方法、应用领域以及最新进展等方面进行详细探讨。
电子结构计算的基本原理
电子结构计算的核心目标是求解多电子体系的薛定谔方程,但由于其计算复杂度极高,通常采用近似方法进行求解。其中,密度泛函理论(DFT)是最广泛使用的电子结构计算方法之一。
DFT通过将多电子体系的电子结构问题转化为单电子问题,利用Hohenberg-Kohn定理,将体系的基态能量表示为电子密度的泛函。这种方法在计算效率和精度之间取得了良好的平衡,适用于从分子到固体材料的广泛范围。
除了DFT,还有其他一些重要的电子结构计算方法,如哈特里-福克方法(Hartree-Fock,HF)和多组态展开方法(Configuration Interaction,CI)。HF方法通过求解自洽场方程,考虑了电子之间的交换相互作用,但忽略了电子相关能,因此在处理复杂体系时精度有限。而CI方法则通过考虑多个电子组态的叠加,能够更准确地描述电子相关效应,但计算成本较高,通常用于小分子体系。
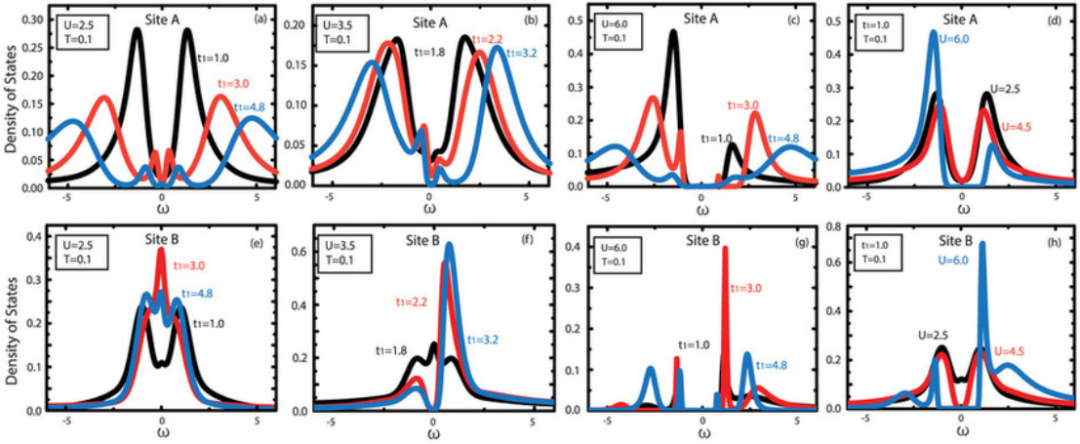

电子结构计算的常用方法
密度泛函理论(DFT)
DFT是目前电子结构计算中最常用的方法之一,其核心思想是通过电子密度来描述体系的基态性质。DFT的计算过程通常包括以下几个步骤:
构建晶体结构:根据实验数据或已知结构,确定材料的原子排列方式。
选择交换关联泛函:常用的交换关联泛函包括局域密度近似(LDA)、广义梯度近似(GGA)和杂化泛函(如HSE06)等。不同的泛函会影响计算结果的精度和适用范围。
设置计算参数:包括平面波基组的截断能、k点网格的密度、赝势的选择等。这些参数的选择对计算结果的收敛性和精度有重要影响。
计算电子结构:通过迭代求解Kohn-Sham方程,得到电子的占据能级、能带结构、态密度等信息。
局域高斯基组方法
局域高斯基组方法是一种基于高斯型轨道的电子结构计算方法,适用于处理大分子体系。该方法通过将电子波函数表示为高斯型轨道的线性组合,能够有效减少计算复杂度,同时保持较高的精度。这种方法在有机分子和生物大分子的电子结构计算中具有广泛应用。
平面波基组与投影缀加波(PAW)赝势
平面波基组是一种常用的电子结构计算方法,特别适用于周期性体系(如晶体和纳米材料)。PAW赝势方法通过将原子核的强相互作用与电子的弱相互作用分开处理,能够有效减少计算中的数值误差,并提高计算效率。
这种方法在第一性原理计算中被广泛采用,尤其是在处理过渡金属和复杂材料时。
半经验方法是一种在计算复杂大分子电子结构时采用的近似方法,通过引入参数化模型来简化计算。这种方法通过估计难以计算的积分,基于电子结构的试验资料,从最简单的模型Hamilton量出发,考虑分子间的相互作用,包含待定参数。
这种方法简化了计算工作量,适用于更复杂分子的电子结构计算,结果具有定性和半定量特性,足以说明分子性质,验证或否定物理化学假设。
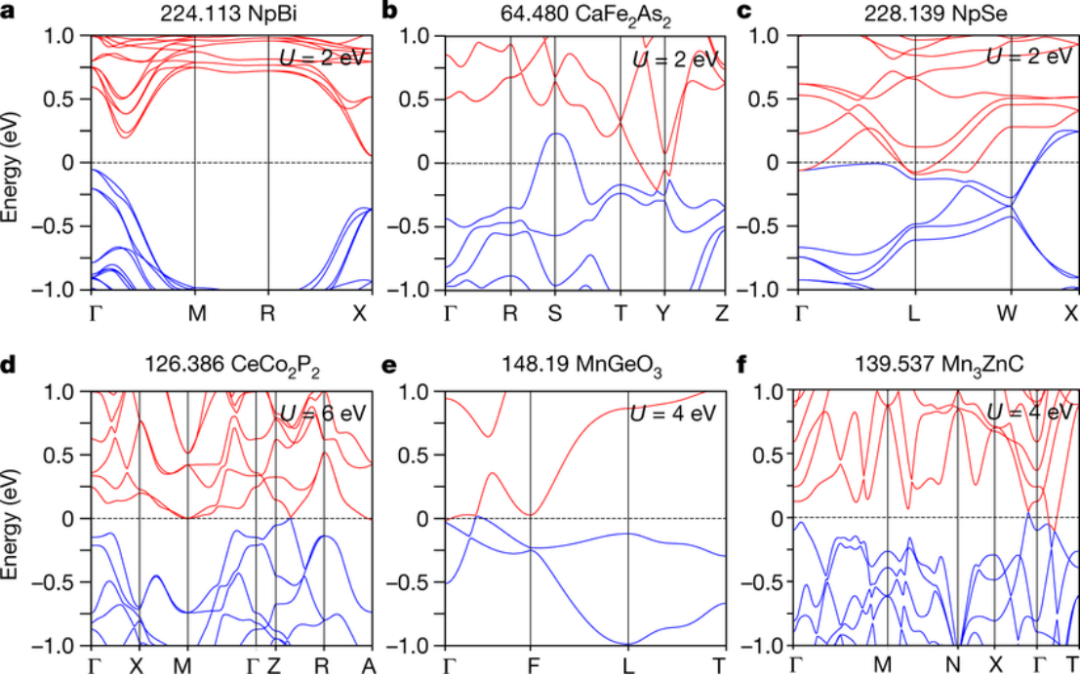
电子结构计算的应用领域
材料科学
电子结构计算在材料科学中的应用非常广泛,包括:
半导体材料:通过计算能带结构和态密度,可以预测材料的导电性质、光吸收特性等,为新型半导体材料的设计提供理论支持。
催化材料:通过计算反应路径和过渡态能量,可以预测催化剂的活性和选择性,为高效催化剂的设计提供依据。
磁性材料:通过计算磁矩和自旋态,可以研究材料的磁性行为,为磁性材料的开发提供理论指导。
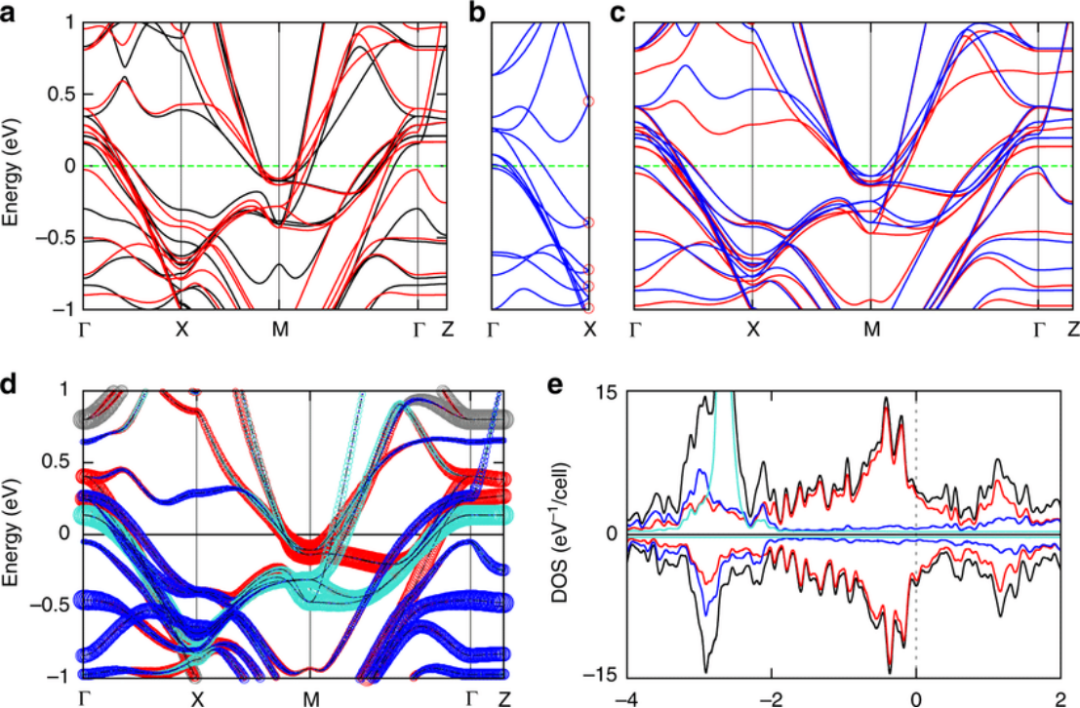
化学
电子结构计算在化学中的应用主要包括:
分子性质预测:通过计算分子的电子结构,可以预测其键长、键角、振动频率等性质,为分子结构的解析提供理论支持。
反应机理研究:通过计算反应路径和过渡态能量,可以揭示化学反应的微观机制,为反应动力学的研究提供理论依据。
物理
电子结构计算在物理中的应用主要包括:
凝聚态物理:通过计算电子的能带结构和态密度,可以研究材料的电子输运性质、磁性行为等,为凝聚态物理的研究提供理论支持。
量子化学:通过计算分子的电子结构,可以研究分子的电子激发态、光谱特性等,为量子化学的研究提供理论依据。
总结
电子结构计算是材料科学、化学、物理等领域中一项基础且重要的研究工具。通过密度泛函理论、局域高斯基组方法、平面波基组与PAW赝势等方法,可以精确预测和分析物质的电子结构。
随着人工智能和高性能计算技术的发展,电子结构计算的效率和精度将进一步提高,为新材料的开发和新理论的提出提供强有力的支持。未来,电子结构计算将在更多领域发挥重要作用,推动科学技术的发展。
【做计算 找华算】