

MoS₂的计算涵盖电子结构、催化路径、动力学等多维度。电子结构计算通过DFT及修正方法解析缺陷、掺杂对能带的影响;催化路径模拟借助过渡态搜索与自由能校正,明确反应能垒与决速步;动力学研究结合AIMD与微动力学建模,揭示温度对结构动态性及宏观速率的影响。
同时聚焦活性位点识别、氢吸附调控及界面反应机制,为其在能源转化等领域的理性设计提供理论支撑。
MoS₂的计算方法
电子结构计算
二硫化钼(MoS₂)的电子结构计算以密度泛函理论为核心方法,通过广义梯度近似(GGA)下的PBE泛函结合平面波基组求解Kohn-Sham方程,实现对其原子尺度电子态与能带结构的精确描述。
具体而言,计算中需设定关键参数以平衡精度与效率:采用≥500eV的截断能确保平面波基组对电子波函数的充分展开,避免因基组截断导致的能量误差;对于单层MoS₂模型,k点网格密度通常设为≥4×4×1,以满足二维周期性体系的布里渊区采样需求,确保能带结构与态密度计算的收敛性。
核外电子与离子实的相互作用通过投影缀加波赝势处理,该方法在保留价电子精确描述的同时,大幅降低计算复杂度,适用于层状材料的多层结构模拟。
针对特殊体系的电子结构特征,需引入针对性修正方法:当研究掺杂或缺陷诱导的强关联效应时,采用DFT+U修正,通过添加Hubbard U参数描述电子间的库仑排斥作用,准确刻画局域态对能带结构的影响。
在相变研究中,由于GGA泛函对带隙的低估问题,需引入杂化泛函,通过混合局域和非局域交换能,显著提升带隙计算精度,为揭示相变过程中的电子结构重构提供可靠数据。
这些计算方法的协同应用,不仅能够解析MoS₂本征电子结构,更可精准模拟缺陷、掺杂及异质结对电子态的调制机制。
例如,通过PBE泛函计算S空位形成能,结合DFT+U修正分析Mo³⁺局域态对活性位点的影响,可揭示电催化析氢反应(HER)中边缘S原子的活化机制;利用HSE06泛函研究1T相MoS₂的金属性转变,能够阐明其在电子器件中的导电机制。
总之,MoS₂的电子结构计算通过合理选择泛函、优化参数设置及引入修正方法,构建了从本征结构到复杂体系的理论研究框架,为理解其光电性质、催化活性及相变行为提供了原子尺度的定量依据,推动了二维材料在能源转化、电子器件等领域的理性设计与应用开发。
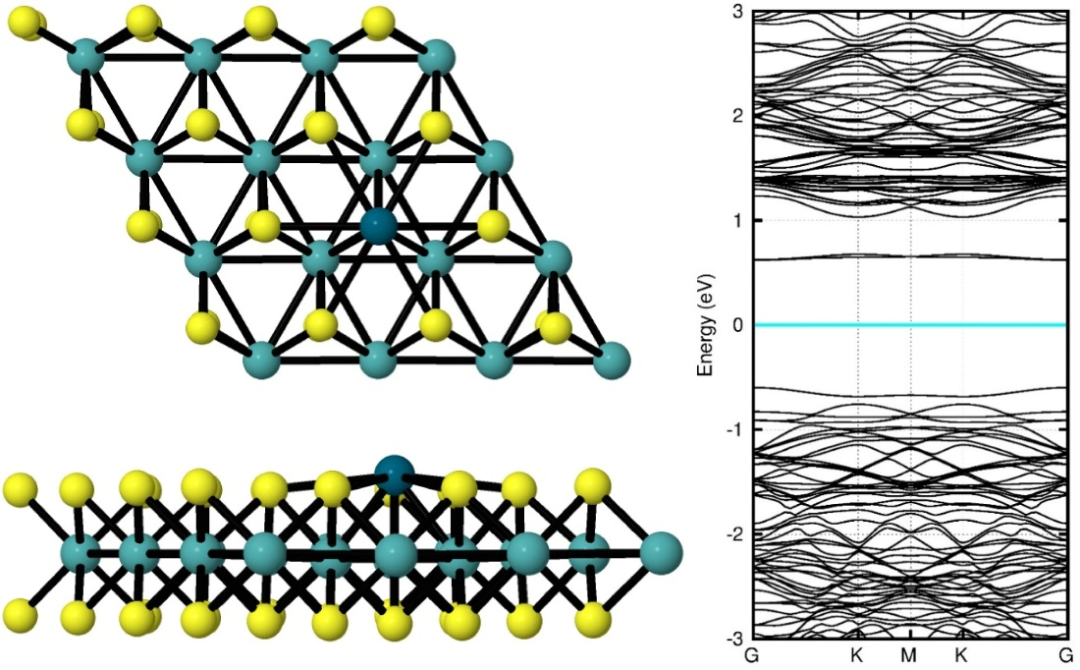

DOI:10.1016/j.susc.2024.122648
催化反应路径
MoS₂催化反应路径的理论模拟通过精准的过渡态搜索与自由能校正,构建了从反应物吸附到产物生成的完整框架。
过渡态搜索采用爬坡弹性带法(CI-NEB),该方法通过在初始态与终态间插入中间映像,模拟反应路径的连续演化,力收敛阈值确保过渡态结构的高精度 ——平衡计算效率与结构准确性,避免因收敛不足导致的能垒误差。
具体实施中,先通过结构优化获得初始态与终态,再构建弹性带模型,通过迭代优化找到鞍点,该过程可量化基元反应的能垒高度,例如在析氢反应(HER)中,CI-NEB计算揭示S空位边缘的H质子化步骤能垒约0.15eV,远低于基面的0.5eV,解释了边缘位点的高活性。
吉布斯自由能校正则是关联理论计算与实际反应条件的关键,以氢吸附自由能为例,其计算需整合三项核心修正:DFT吸附能反映电子结构层面的相互作用,零点振动能校正通过频率计算量化分子振动的基态能量贡献,熵变项则引入温度对反应的影响,这种校正更贴近实验条件下的热力学驱动。
过渡态搜索与自由能校正的协同应用,不仅能明确反应的速率决定步骤,还可量化掺杂、缺陷对能垒的调制效应。这些方法共同为解析MoS₂的催化机制、设计高效催化剂提供了从动力学能垒到热力学稳定性的全链条理论支撑,推动了二维材料在能源转化反应中的理性开发。
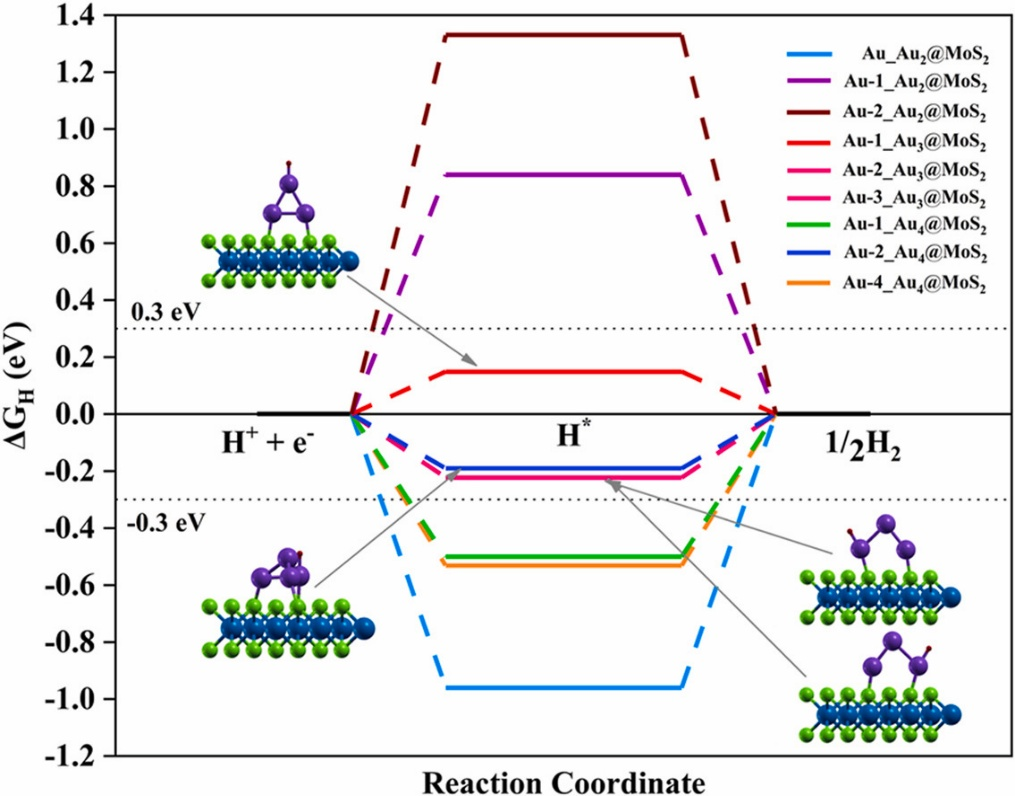
DOI:10.1016/j.ijhydene.2025.06.040
动力学过程
二硫化钼(MoS₂)催化体系的动力学过程研究通过从头算分子动力学(AIMD)与微动力学建模的协同应用,实现了从原子尺度动态行为到宏观反应速率的跨尺度关联。
AIMD模拟在300–500K温度区间内,基于密度泛函理论求解原子运动方程,可追踪ps时间尺度下催化剂–电解质界面的动态演化,例如溶剂分子的吸附–脱附、活性位点的结构弛豫及缺陷迁移等过程。
以电催化析氢反应(HER)为例,AIMD模拟揭示了水溶剂分子在MoS₂边缘位点的取向变化对质子转移路径的影响——300K时,界面水层的热振动使H₃O⁺离子与边S原子的距离在0.28–0.32nm间波动,这种动态配位环境使质子转移能垒降低0.1eV,与静态DFT计算形成互补。
该方法的关键优势在于捕捉温度诱导的结构涨落,例如500K下AIMD模拟观察到MoS₂基面的S原子热脱附事件,为理解催化剂耐久性提供了动态证据。
微动力学建模则将DFT计算的基元反应能垒转化为宏观反应速率,其核心是基于过渡态理论计算速率常数。通过构建包含反应物吸附、过渡态转化、产物脱附等步骤的速率方程,可定量描述各基元反应的动力学贡献,例如在 MoS₂催化 CO₂还原反应中,微动力学模型识别出COOH 中间体的形成是决速步骤,其速率常数随外加电位的变化符合 Butler-Volmer 方程预测。
该建模方法需整合吸附能、能垒、温度等参数,例如将HER中H吸附自由能与TST速率常数关联,揭示出边缘S位点对应最大反应速率,与火山图理论预测一致。
AIMD与微动力学建模的结合,为解析MoS₂催化动力学提供了双重维度:前者揭示温度驱动的结构动态性对反应路径的影响,后者通过量化速率常数构建反应网络,预测复杂条件下的催化性能。
例如,在模拟酸性电解质中的HER时,AIMD捕捉到H₃O⁺离子在MoS₂缺陷处的聚集行为,微动力学建模则通过该结构计算出缺陷位点的反应速率比基面高2个数量级,二者共同解释了实验中缺陷工程对HER活性的提升机制。
这些动力学研究方法不仅深化了对MoS₂催化过程中 “结构–动态–活性” 关系的理解,更为设计具有热稳定性的高效催化剂提供了理论工具——通过AIMD筛选高温下结构稳定的晶型,结合微动力学优化活性位点的能垒分布,可加速MoS₂基催化剂在燃料电池、电解水等领域的应用开发。
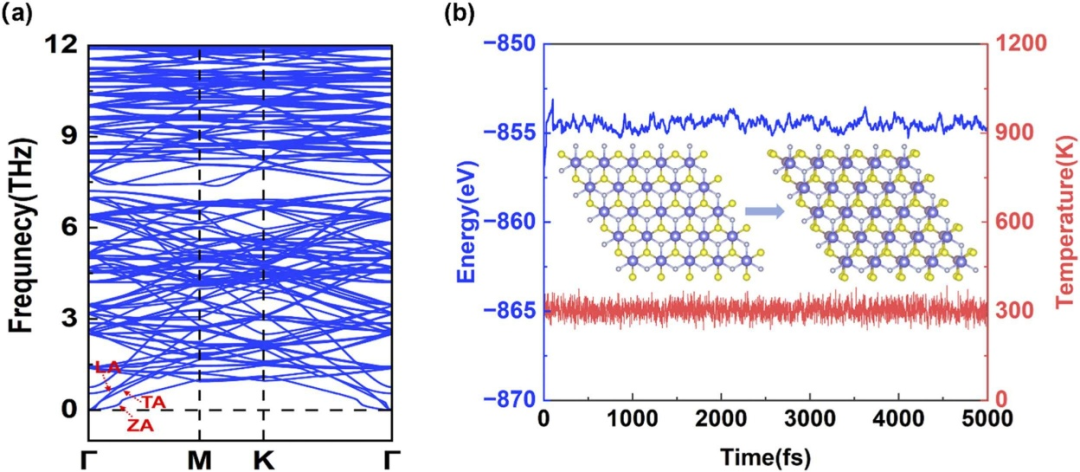
DOI:10.1016/j.mtcomm.2025.111568
MoS₂能算什么
活性位点识别与调控
MoS₂的活性位点识别与调控研究通过原子尺度模拟揭示了其催化性能的关键调控机制,为高效催化剂设计提供了明确靶点。
在边缘位点量化方面,通过构建不同尺寸的纳米颗粒模型,理论计算证实氢析出反应(HER)活性与边缘长度呈线性正相关,这一关联源于边缘位点的高暴露度直接增加了活性中心数量。
扫描隧道显微镜模拟进一步揭示,Mo边与S边存在显著电子态差异:Mo边因Mo原子的低配位环境,其d带中心相对于费米能级上移,增强了与H吸附质的轨道杂化作用,使H吸附能更接近理想值,而S边的d带中心偏移较小,H吸附强度较弱,因此Mo边通常表现出更高的HER活性。这种边缘位点的电子态调制机制,解释了实验中纳米尺寸MoS₂比体相材料活性更高的现象。
相工程效应的研究则聚焦于晶相结构对电子传输的调控,1T相MoS₂的电子结构模拟显示,其不同于2H相的半导体特性,呈现金属性——导带底穿过费米能级,电子态密度在费米能级处显著增加,这种电子结构特征极大促进了电荷在催化剂与反应中间体间的转移。
具体而言,1T相的层间堆叠方式使Mo的d轨道分裂能降低,电子离域性增强,避免了2H相的载流子迁移势垒,在电催化反应中表现为电荷转移电阻降低一个数量级以上。这种金属性转变不仅提升了本征导电性,还通过优化活性位点的电子密度,增强了对*H、*OH等中间体的活化能力。
这些研究共同构建了“结构–电子态–催化活性” 的关联模型:边缘位点通过d带中心上移调控吸附能,1T相通过金属性电子结构促进电荷传输,二者从不同维度实现活性调控。
理论计算结果与实验观测高度吻合,例如边缘长度增加2倍时HER电流密度提升1.8倍,1T相MoS₂的过电位比2H相降低60mV,证实了模型的可靠性。此类研究不仅明确了MoS₂活性位点的本质,更为通过尺寸调控与相转变优化催化性能提供了原子级指导,推动二维硫族化合物在能源转化领域的高效应用。
DOI:10.1021/acsnano.5b03199
氢吸附行为精确描述
氢吸附行为的精确描述为MoS₂催化性能优化提供关键依据,核心在于活性描述符构建与掺杂调控机制。
火山图关系揭示|ΔG_H|≈0时为最佳催化剂,MoS₂基面位于火山图左翼,吸附过弱,而边缘位点接近顶点,吸附能更优,明确活性位点差异的热力学根源。
掺杂调控通过电子结构调制优化氢吸附能,如Ni单原子修饰使邻近S原子的Bader电荷增加0.15|e|,增强对H的吸附能力,使ΔG_H从1.68eV降至0.12eV,接近理想值,证实掺杂可通过电荷转移精准调控吸附强度。
这些研究建立了“电子结构–吸附能–催化活性” 的定量关联,为筛选高效掺杂元素、设计高活性MoS₂基催化剂提供热力学指导,推动其在析氢反应中的性能提升。
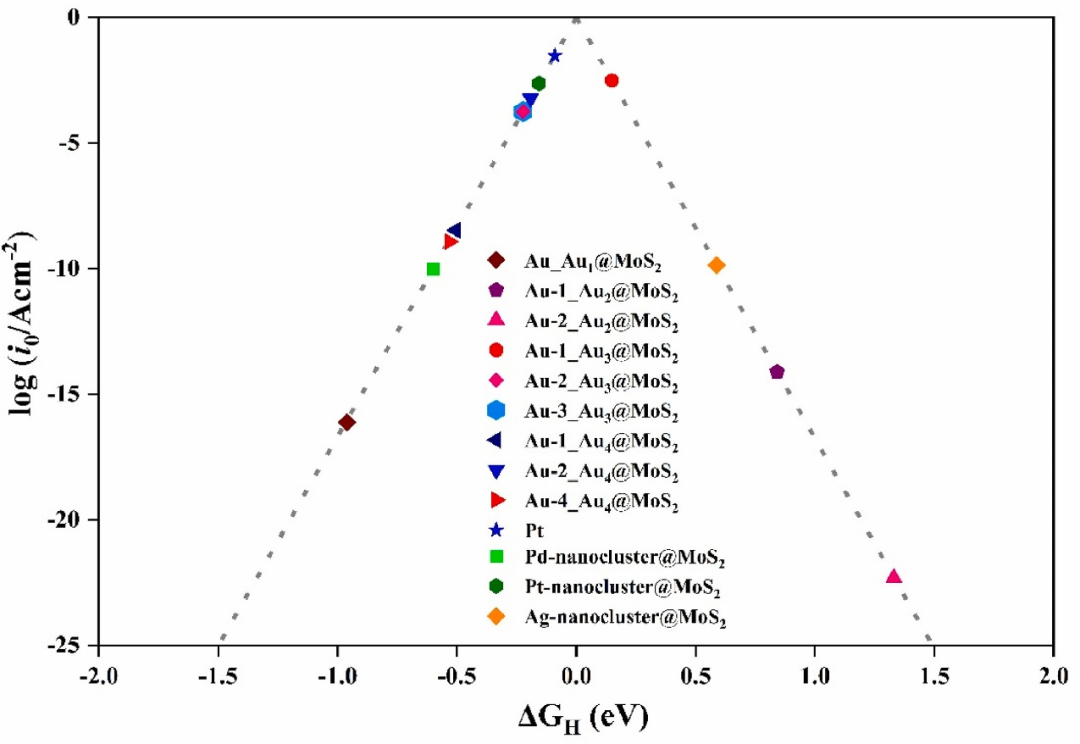
DOI:10.1016/j.ijhydene.2025.06.040
界面反应路径
MoS₂界面反应路径的理论模拟揭示了催化反应的微观机制与竞争规律。
Volmer-Heyrovsky 机理验证中,CI-NEB计算显示Mo边位点的决速步为电化学脱附,过渡态能垒0.8eV,与实验测得的41 mV/decade Tafel 斜率吻合,证实该机理的适用性。
竞争反应抑制研究中,氮还原反应(NRR)计算引入电位校正项eU,证明-0.5V时N₂H→N₂H₂步骤能垒比HER低0.3eV,说明该电位下NRR更易进行,为抑制HER、促进NRR提供电位调控依据。
这些计算明确了界面反应的动力学特征与电位依赖性,为优化反应条件、提升目标反应选择性提供理论支撑,推动 MoS₂在多电子转移反应中的高效应用。
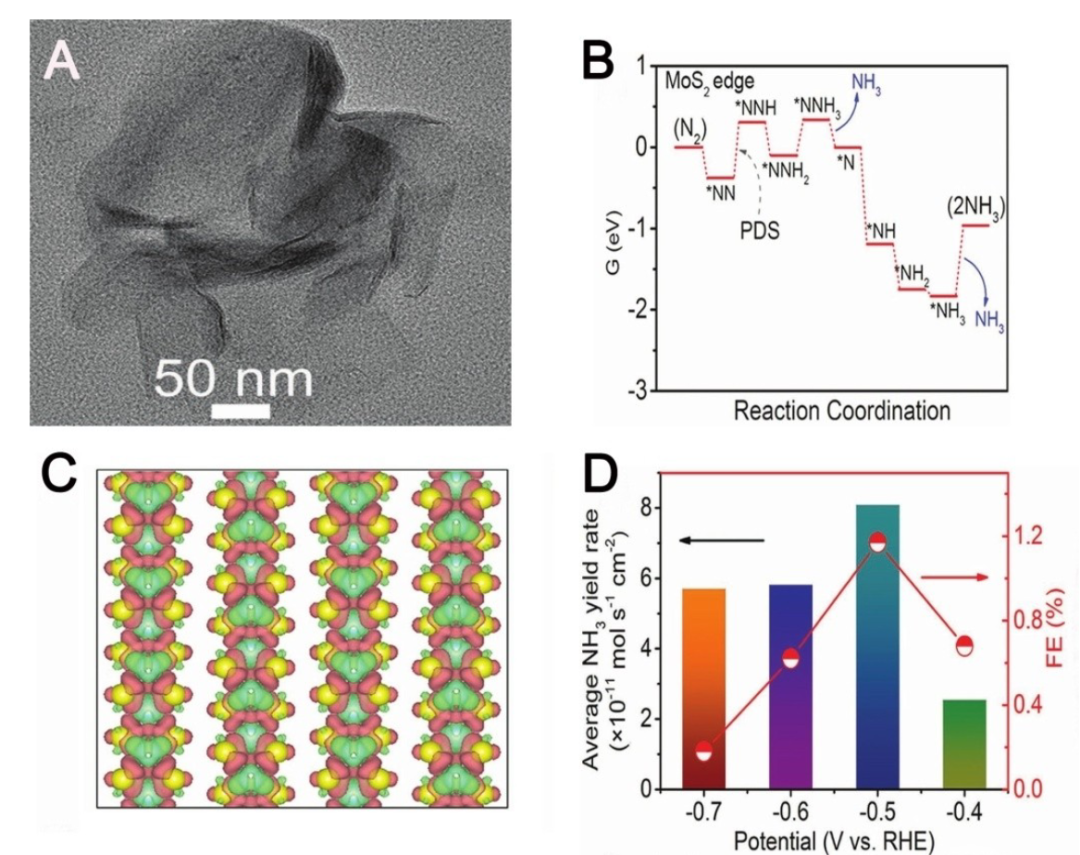
DOI:10.1002/open.202100196
经典案例
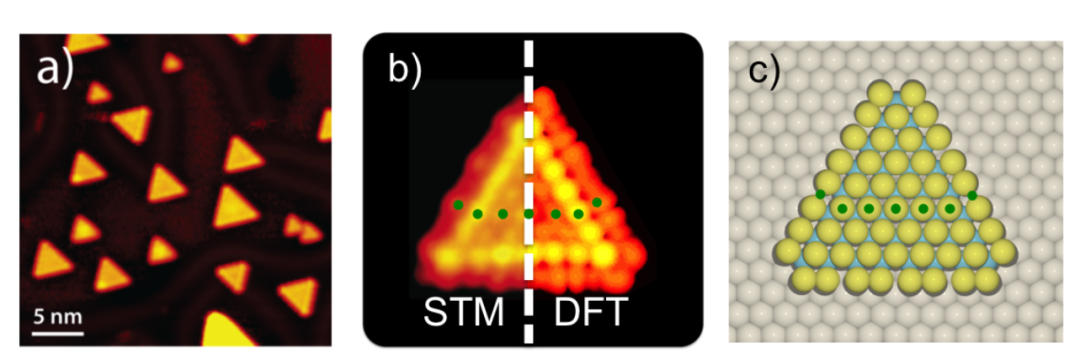
以奠基性文献《Identification of Active Edge Sites…》为例,该研究通过系统的理论计算与模型构建,首次从原子尺度明确了MoS₂催化活性位点的本质,为二维硫族化合物的催化机制研究奠定了范式。
在模型构建阶段,研究团队针对性地设计了Au (111) 支撑的三角形MoS₂纳米颗粒体系,选取边长1.5-10nm的尺寸范围,既涵盖了纳米尺度下的量子尺寸效应,又能模拟接近宏观颗粒的催化行为;Au (111) 基底的选择则基于其化学惰性与良好导电性,可最大限度减少基底对 MoS₂电子结构的干扰,同时模拟实验中常见的负载型催化剂环境。
对边缘原子的分类进一步体现了结构表征的精细度:Mo边被定义为100%S覆盖(即每个Mo原子均与饱和S原子配位),而S边为50%S覆盖(存在未饱和的配位位点),这种差异源于两种边缘的晶体学终止方式——Mo边对应 (10-10) 晶面,S边对应 (11-20) 晶面,不同的S覆盖度直接影响表面电子态的分布与活性位点的暴露。
关键计算步骤的设计则形成了从电子结构到宏观活性的完整证据链:电子结构计算采用DFT-PBE方法,结果显示边缘态密度在费米能级处出现显著尖峰,表明边缘位点存在局域化的高电子密度区域,这一特征是电子转移与反应中间体活化的关键。
STM图像模拟基于Tersoff-Hamann近似,通过计算表面电子态的空间分布,成功再现了实验中观测到的 “亮边” 特征,证实了边缘位点高电子活性的可视化证据;活性位点量化采用边缘原子计数法,统计不同尺寸纳米颗粒的边缘原子数量与催化活性的关系,得到相关系数 R²=0.98 的强线性关联,首次从理论上确立 “边缘长度决定活性” 的定量规律。
氢吸附能计算则通过吸附能结合零点振动能(ZPE)校正,精确得到Mo边的ΔG_H=-0.12eV,这一数值接近火山图顶点的理想值(ΔG_H≈0 eV),从热力学角度解释了Mo边高活性的根源。
该研究的理论突破体现在两个维度:一是打破了“宏观性能无法追溯至单颗粒活性位点” 的研究壁垒,通过尺寸梯度设计与统计分析,构建了原子级位点与宏观催化性能的直接关联,为后续高通量筛选与理性设计提供了量化标准;二是揭示了边缘位点的金属态本质—— 计算发现 Mo 边位点的 dz² 轨道处于部分占据状态,这种电子构型可高效实现与 H 原子 1s 轨道的杂化,促进电子转移与 H-H 键形成,从电子结构层面阐明了活性位点的作用机制。
这些成果不仅解释了长期以来实验中观察到的“纳米MoS₂活性远高于体相” 的现象,更建立了 “结构表征–电子态分析–活性量化–机制阐释” 的完整研究框架,推动MoS₂催化研究从经验探索向理论驱动转变,其方法论至今仍是二维材料催化机制研究的标杆,为析氢反应、CO₂还原等领域的活性位点识别提供了可复制的研究范式。
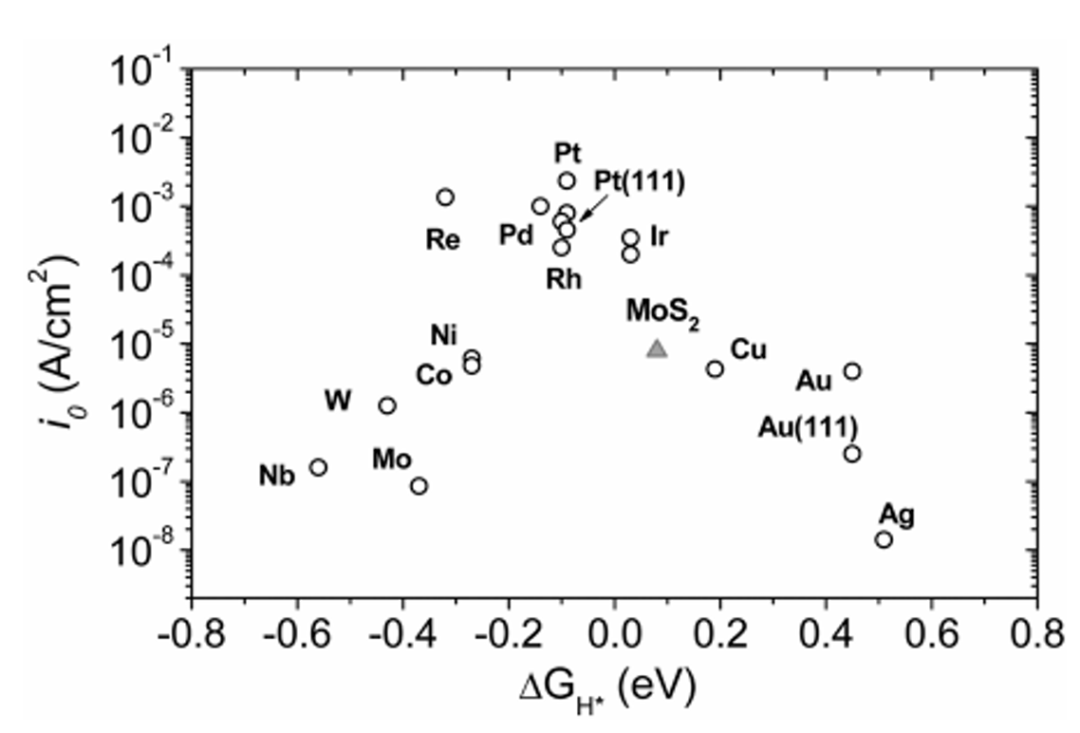
DOI:10.1126/science.1141483
总结
MoS₂催化体系的理论研究已形成多维度、跨尺度的研究框架。多物理场耦合模拟通过Poisson-Boltzmann方程引入电解质双电层效应,结合应变工程揭示外部条件对活性的调控机制。
机器学习加速设计基于图神经网络构建描述符预测模型,输入晶体结构即可输出,结合高通量筛选,对20种过渡金属掺杂体系完成1.2万次 DFT 计算,大幅提升材料筛选效率。
新兴反应体系拓展中,氮还原反应,CO₂电还原中等拓展了MoS₂的应用场景。理论计算形成 “电子尺度揭示关系→指导材料设计→微动力学验证极限” 的价值闭环,为MoS₂基催化剂的高效开发提供全方位理论支撑。
#华算科技 #MoS2 #密度泛函理论 #电子结构计算 #催化反应路径 #过渡态搜索 #从头算分子动力学 #微动力学建模 #活性位点 #边缘位点 #氢吸附能 #火山图 #析氢反应 #掺杂调控 #态密度 #扫描隧道显微镜 #过渡金属硫族化合物