

 说明:本次主要介绍机器学习辅助的X射线吸收光谱(XAS)分析在双金属催化剂研究中的应用。文章介绍了双金属纳米粒子(BNPs)的特性、X射线吸收精细结构(XAFS)光谱技术的应用和局限性,及机器学习辅助XAFS分析在双金属催化剂结构表征中的应用。想要了解更多相关知识请参考以往内容:
说明:本次主要介绍机器学习辅助的X射线吸收光谱(XAS)分析在双金属催化剂研究中的应用。文章介绍了双金属纳米粒子(BNPs)的特性、X射线吸收精细结构(XAFS)光谱技术的应用和局限性,及机器学习辅助XAFS分析在双金属催化剂结构表征中的应用。想要了解更多相关知识请参考以往内容:什么是同步辐射R空间?
精选干货|同步辐射PDF基础知识及经典应用分析!
双金属纳米颗粒(BNPs)由两种不同金属组成,其独特性能使其在催化领域备受关注。与单金属相比,BNPs的尺寸、形状和结构赋予其优异的光学、电子、磁性、热和催化性能。
这些纳米颗粒可集成到纳米复合材料中进一步提升性能,且因其尺寸小、表面积大,在催化应用中表现出色。
近年来,研究者致力于开发新型BNPs,如合金、核壳结构和金属间化合物。双金属催化剂在能源转换和化学转化中表现出色,通过添加第二金属可调节其催化性能。
BNPs可分为合金、金属间和纳米复合结构三类,其催化性能可通过几何效应或电子效应进行调控。
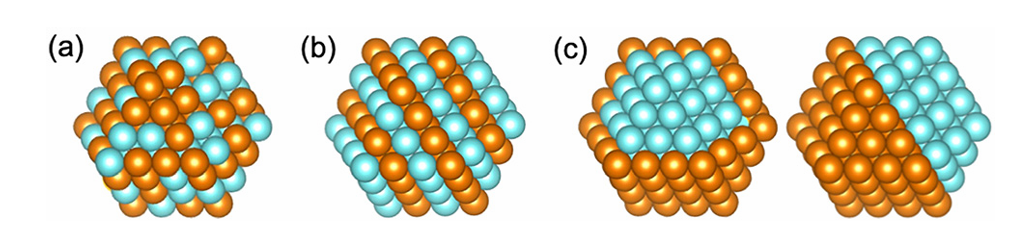
图1. 三种常见双金属结构的示意图:合金(a)、金属间化合物(b)以及两种可能的纳米复合材料变化(c)。
XAFS光谱学是研究BNPs在实际反应条件下原子尺度特征及其与催化性能关联的有力工具。然而,XAFS存在信噪比低和对表面物种灵敏度有限的局限。机器学习(ML)在XAFS光谱结构精炼中的应用为解决这些挑战提供了新途径。
下面将介绍ML在XANES和EXAFS光谱中的应用。最初将监督式机器学习应用于单金属纳米颗粒的XANES光谱的主要思想是提取关键的结构特征,即成对配位数(CNs)。
这些数据可以用作纳米颗粒尺寸、形状和形态的指标,但由于上述的局限性,EXAFS难以测量这些数据。
对于双金属纳米颗粒(BNPs),部分配位数(CNs)被用来提取上述的组成信息。在这两种情况下,人工神经网络都是在一个理论数据集上进行训练,该数据集包含使用FEFF和FDMNES理论光谱学代码生成的数十万光谱。
通过使用这两种代码进行神经网络训练,可以部分补偿其中存在的一些系统误差。
2.1 机器学习辅助的XANES分析
对于单金属纳米颗粒(NPs),训练集(标签为几个最近邻壳层的配位数(CNs))是通过组合方法构建的。
在这个方法中,理论光谱μi(E) 是通过对 n 个随机选择的、特定位置的XANES计算序列 i 进行平均得到的,并且可以用明确定义的、位置平均的CNs进行标记:
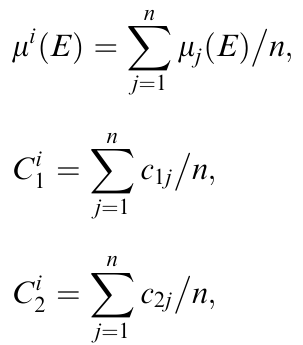
这里,c1j、c2j等是序列 i 中位置j 的第一、第二等邻近壳层的CNs。
对于双金属材料,每种原子类型的XAS光谱边区可提供四种第一近邻对信息:CAA、CAB、CBA和CBB。
获取配位数(CNs)的方法依赖两个独立的、特定于吸收体的神经网络,每个网络输出特定对的信息(如吸收体A对应CAA和CAB,吸收体B对应CBA和CBB)。神经网络通过线性组合特定位置的理论XANES光谱进行训练。
为减少NN预测的系统误差,研究者采用多种方法,如利用纳米颗粒与块状光谱差异进行预测、重复训练估计误差范围、使用神经网络集成(NNE)量化不确定性,以及通过迁移学习提升预测质量。
NN的非线性特性使其在处理系统误差和实验噪声时优于线性方法。

图2:NN-XANES应用于AxB1-x双金属系统的示意图。从A和B吸收组分的XANES中提取部分第一配位数(CNs)。部分CNs(A-A、A-B、B-B和B-A)用于推断平均纳米颗粒结构。
2.2 ML-XANES方法在BNP催化剂结构表征中的应用案例
使用 ML-XANES 方法为 BNPs 的结构表征和建模开辟了新路径,其核心依据是配位数(CNs),以往这多局限于 EXAFS 分析。理论上,部分 CNs(CAA、CAB、CBA、CBB)能从 ML-XANES 分析中在整个组成范围内获取。
在稀释金属负载、苛刻反应条件以及 BNPs 高结构无序等情况下,这相比 EXAFS 具有独特优势,因为此时 EXAFS 数据可能质量极差或无法获取,例如平面载体上尺寸选择性的双金属簇。
在这种情形下,ML-XANES 成为唯一可量化邻近对的工具,如 Cu₃Pd 和 Cu₄Pd 双金属簇所示。
当构成元素在周期表中相邻,导致Z 对比度较差时,EXAFS 建模难以从数据中提取部分 CNs(尽管此时仍可获得对应于 A-M 和 B-M 对的 CNs,其中 M = A 或 B)。
而ML-XANES 对这些光谱特征敏感,并且因其对合金原子间电荷转移的强烈敏感性,这些特征会受不同邻近类型对 X 射线吸收体的 CNs 的显著影响。
近期,在相关研究中,展示了ML-XANES 在分析 Pt-Au BNPs 以及确定用于结构建模的 CNs 方面的强大功能。
该方法融合了ML 辅助的 XAFS 拟合,用于结构和组成分析。构建了NN 以确立 XANES 光谱与第一近邻对 Au-Au、Au-Pt、Pt-Au 和 Pt-Pt 的 CNs 之间的关联。图 3 的 NN 验证结果表明,其可根据理论 XANES 光谱对部分 CNs 进行良好预测。
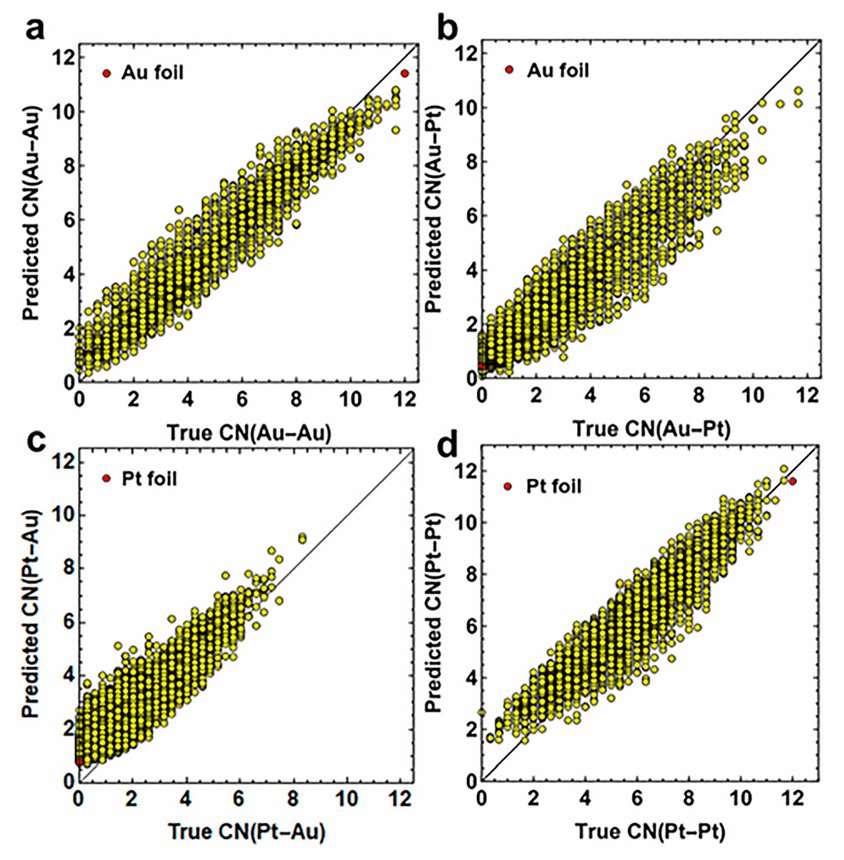
图3:通过预测的(a)CN Au-Au、(b)CN Au-Pt、(c)CN Pt-Au和(d)CN Pt-Pt与真实值对比验证NN。
(ACS Appl. Nano Mater., 2022, 5, 8775–8782.)
过去十年,稀释物种(如Pt、Ag、Pd和Rh)的BNPs因提升选择性及与宿主(如Au、Ag)的协同效应,在催化领域备受关注。
近期研究聚焦预处理对稀释型(4-8原子% Pd-in-Au)BNPs催化HD交换反应活性的影响。通过催化活性测量、机器学习光谱分析和第一性原理动力学建模,发现活性物种为少数(1-3个)表面Pd集成。
XANES数据显示,经不同预处理(S0-S4),8原子% Pd-in-Au样品的Pd K边吸收系数相对Pd块状参考光谱发生正负偏移。
其中,O₂处理后吸收系数质心向高能移动,氢气处理后向低能移动。正偏移表明Pd偏聚形成(PdₙAu),负偏移则与Pd溶解致Pd-Au邻接增多相关。

图4:多尺度方法(理论计算与实验结合)用于优化纳米多孔稀薄合金催化剂的性能
(Chem. Rev., 2022, 122, 8758–8808)
利用 NN-XANES 方法获取了部分 CNs(CPd-Pd 和 CPd-Au)。图 5b所示趋势与基于图 5a的定性观察相符,即 Pd-Pd CNs 在最初 O₂ 处理后略有增加,后续 H₂ 处理后减少(Pd-Au 增加)。
比较 EXAFS 分析和 NN-XANES 分析结果可知,后者更优,因为 EXAFS 对稀释元素的最近邻 CNs(如 Pd-Pd 与 Pd-Au)灵敏度欠佳。
相比之下,NN-XANES 方法表现出色,神经网络对 CN 预测所需的各种特征具有 “敏锐洞察力”,这相较于依赖单一标准的EXAFS 数据分析中使用的标准最小二乘拟合方法(仅最小化均方误差)是一大改进。
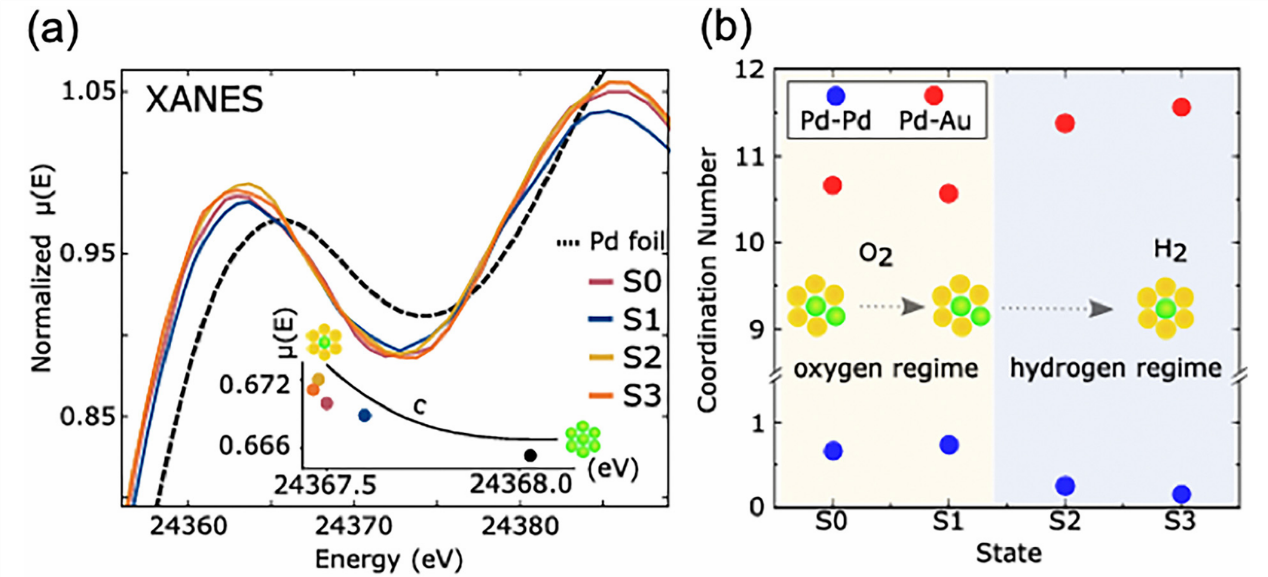
图5:不同预处理后稀释型(8原子% Pd在Au中)BNPs样品收集的归一化XANES(a),以彩色条带显示为“O₂模式”和“H₂模式”(b)。(a)图的插图显示了光谱质心“c”的偏移。
来自NN-XANES的CNs与Pd原子在O₂处理后具有更多Pd邻近原子、在H₂处理后具有更少Pd邻近原子一致。(a)和(b)图中的原子Pd集成以俯视图示意图表示(黄色=Au;绿色=Pd)。
(Nat. Commun., 2022, 13, 832.)
2.3 机器学习辅助的EXAFS分析
BNPs的EXAFS数据质量良好时,能提供X射线吸收原子最近邻原子结构和动力学的丰富信息,远超XANES。
确定EXAFS信息内容的方法(如奈奎斯特准则依赖方法)已使用半个世纪。例如,对于同类型纳米颗粒样品,EXAFS光谱的k范围从2到20 Å⁻¹,拟合范围在r空间从2到6.2 Å,约有60个相关独立数据点,可使用多达59个拟合变量,描述局部环境直到第五配位壳层。
而NN-XANES只能确定约四个参数。EXAFS数据的正确解释依赖于X射线吸收原子周围邻居的径向分布函数(RDF)是准高斯的关键假设的有效性。当无序性强烈不对称时,标准方法失效,而基于监督式机器学习的替代方法可以高效替代。
对于双金属合金(AₓBᵧ),理论EXAFS方程用未知的g(r)=dN/dr表示,A-B单散射路径的EXAFS如下:
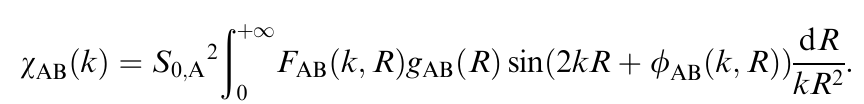
这里,FAB(k,R) 和 ϕAB(k,R) 是理论上可计算的散射幅度和相位函数,S0,A2 是幅度衰减因子。因此,使用通过方程(4)构建的理论训练集,可以在实验EXAFS数据上直接预测未知分布(gAB(R)、gAA(R)、gBB(R)、gBA(R)),而无需事先假设它们的功能形式。
之后,通过在相应的配位壳层范围内积分相应的RDF,可以计算不同类型邻近对(例如,A-B)的CNs和原子间距离(即,在gAB(R)的第(i−1)个和第i个最小值之间):
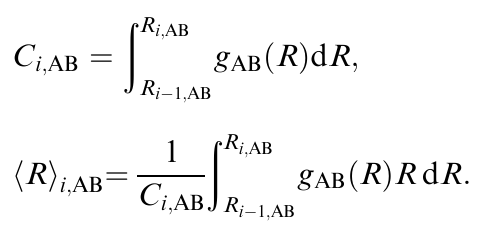
由于方程(4)的反演是一个病态问题,因此使用“真实情况”验证结果分布非常重要,例如将它们与使用分子动力学模拟生成的分布进行比较。
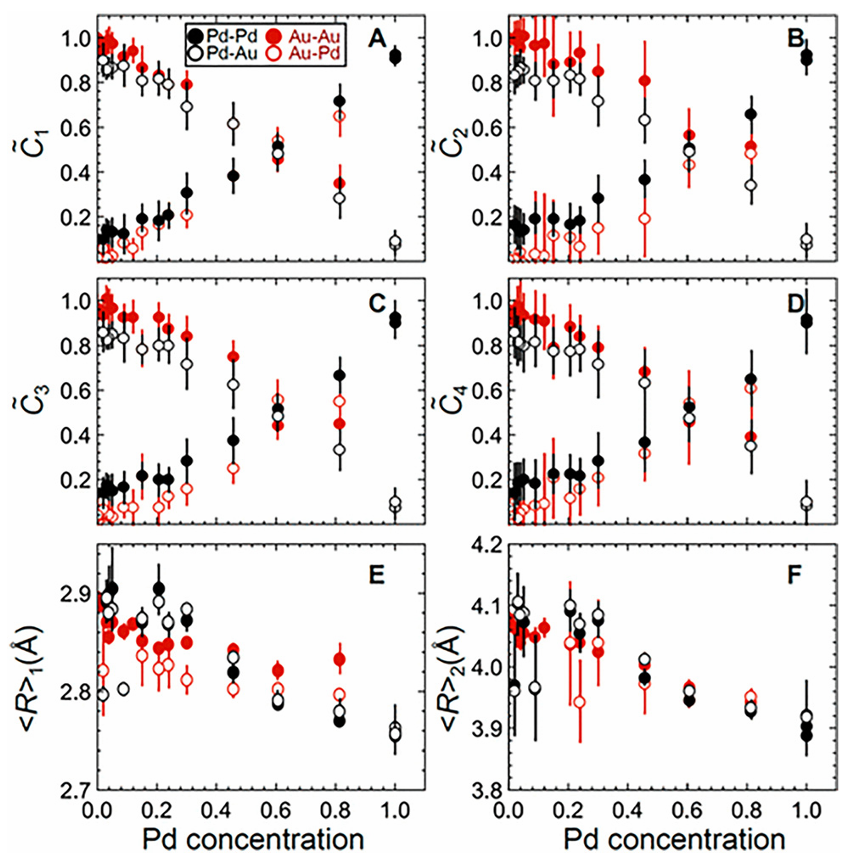
图6:通过NN从实验EXAFS数据中提取的配对径向分布函数(PRDFs)积分得到的前四个配位壳层(a)–(d)的标准化CNs的浓度依赖性,以及前两个配位壳层(e)和(f)的平均原子间距R。
(Chem. Sci., 2020, 11, 3727–3736.)
2.4 ML-EXAFS方法在BNP催化剂结构表征中的应用示例
首次将NN-EXAFS方法应用于BNPs分析的是对Pd–Au颗粒的研究。图7a–f分别显示了相应的CNs和原子间距离。
如图7f所示,对于第二配位壳层,Pd–Pd和Au–Au距离的浓度依赖性几乎相同,但Pd–Au对的距离意外地短,表明在低Pd浓度下Pd向BNP表面偏聚。这一结论通过传统的EXAFS分析是不可能获得的。
NN-EXAFS方法被用于研究电化学还原CO₂的Cu–Zn纳米催化剂,帮助理解双金属结构在反应条件下的演变,尤其是Z对比度低时的情况。
该方法通过分析Cu和Zn K边的EXAFS数据,提取了不同样品在制备态和CO₂RR条件下的Cu–O、Cu–M和Zn–O、Zn–M径向分布函数(RDFs)。结果揭示了CuZn纳米颗粒在CO₂RR条件下逐渐的Cu–Zn合金化过程及从紧密堆积到更无序结构的转变。
传统的NN-EXAFS方法依赖分子动力学模拟训练神经网络,而最近提出的“ONNE”方法则不依赖分子动力学模拟,已用于训练NN和反演熔盐中过渡金属和锕系配合物的EXAFS光谱。

图7:从实验Cu K边EXAFS中提取的Cu-O和Cu-M(M=Cu或Zn)径向分布函数(RDFs)。
(Chem. Sci., 2020, 11, 3727–3736)
随着纳米粒子的复杂性增加,包含五种或更多不同元素组分时,粒子内不同原子的自分离、粒子大小和形状的变化等,需要更先进的光谱表征方法。
为了理解材料的功能,特别是对于需要纳米粒子表面精确原子排列的催化作用,对这种结构的原子级理解是至关重要的。
机器学习和神经网络X 射线吸收近边结构(NN-XANES)分析的进步,为识别这些一直以来使用传统表征方法难以实现的关键结构因素打开了大门。