

四氧化三铁(Fe₃O₄)的催化计算研究通过密度泛函理论(DFT)解析其电子结构(如半金属性、铁磁性)、缺陷/界面效应及催化机理(如NEB法计算反应能垒)。
复合材料设计中,异质结构(如Fe₃O₄@MoS₂)通过界面电荷转移与协同效应优化催化活性。未来结合机器学习、极端条件模拟及动态载流子追踪,可拓展其在能源转化与极端工况中的应用,推动铁基材料理性设计。
电子结构与磁性分析
四氧化三铁(Fe₃O₄)的理论计算研究聚焦于其电子结构与磁性特征的原子级解析。
通过密度泛函理论(DFT)计算态密度(DOS)与能带结构,可揭示其半金属性及铁磁性的微观起源:费米能级附近显著的电子态分布表明其导电特性,而B位Fe³⁺离子的d轨道态密度呈现尖锐峰值(如-1.5 eV处),主导磁性贡献,与A位Fe²⁺的宽化态密度形成对比。
自旋极化计算进一步解析了不同自旋方向(↑和↓)的电子分布差异,例如B位Fe³⁺自旋向上态在费米能级附近的占据主导地位,导致净磁矩积累(约4 μB/Fe),结合超交换作用与双交换机制,解释了Fe₃O₄的高居里温度(约850 K)和铁磁有序性。
这类计算不仅量化了A、B位铁原子的局域磁矩差异(如B位4.0 μB vs. A位3.5 μB),还通过能带劈裂(如导带自旋极化率>80%)阐明其半金属特性在自旋电子学器件中的应用潜力,为磁性材料的理性设计与性能优化提供了关键理论依据。
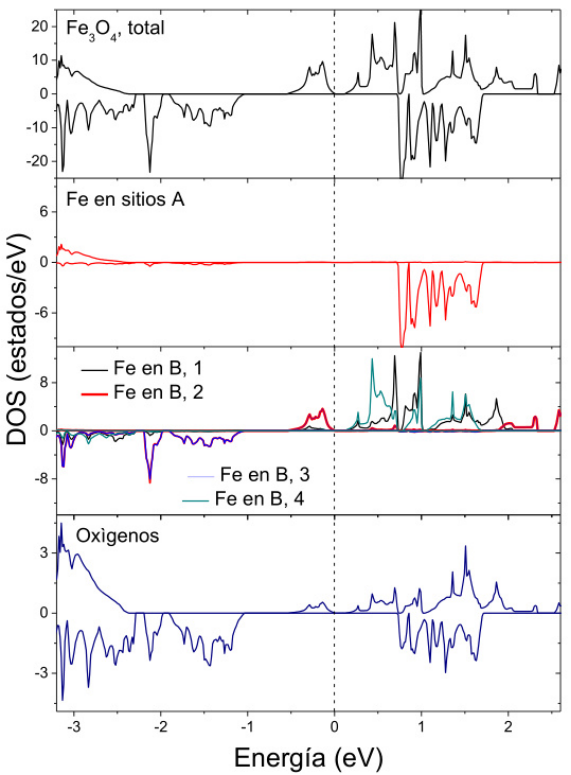

DOI:10.35537/10915/160506
缺陷与界面效应
四氧化三铁(Fe₃O₄)的缺陷与界面效应计算聚焦于微观结构对磁性与催化性能的调控机制。
反位相边界(APB)缺陷的密度泛函理论(DFT)模拟表明,APB-I型缺陷因极低形成能(如-0.3 eV/Ų)广泛存在于晶界,其面状畸变破坏Fe³⁺-O-Fe²⁺超交换作用,导致净磁矩下降(如从4.2 μB降至2.8 μB);
而APB-II型缺陷则通过局部晶格扭曲引入反铁磁耦合(如相邻Fe位点自旋反向排列),降低材料整体铁磁有序性。
表面重构研究揭示不同终止结构的稳定性差异:Fe-O终止表面因氧原子的高电负性形成稳定表面态,而纯Fe终止层在富氧环境中易氧化,动态平衡受表面能(如1.2 J/m²)与吸附能(如O₂吸附能-1.5 eV)共同调控。
表面电子态分析显示,Fe-O终止层的Fe³⁺位点在费米能级附近呈现高态密度(~8 states/eV),为CO氧化或析氧反应(OER)提供活性位点,而纯Fe层则因电子离域化特性更利于电荷传输。
这类计算从缺陷热力学与界面电子结构维度,为磁性器件的性能优化与催化材料的设计提供了原子尺度指导。
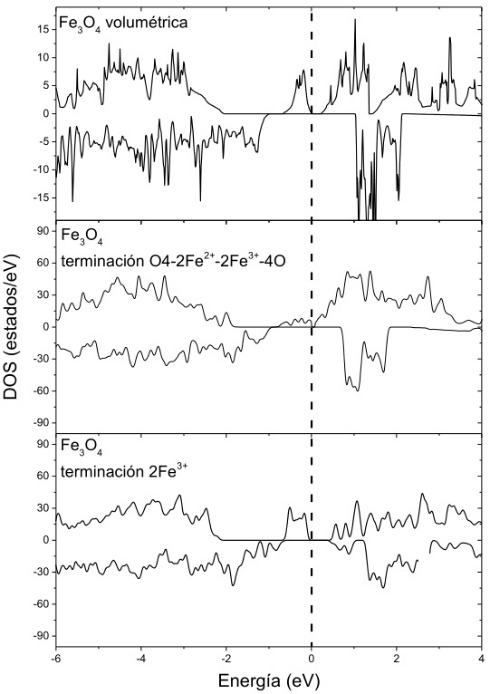
DOI:10.35537/10915/160506
催化反应机理模拟
四氧化三铁(Fe₃O₄)的催化反应机理模拟通过密度泛函理论(DFT)从原子尺度解析其表面活性位点与反应路径的动态关联。
以芳胺化反应为例,Fe₃O₄表面Fe³⁺位点通过电荷转移促进关键中间体[NH₃]⁺的形成,结合微动弹性带(NEB)方法计算的过渡态能垒(如0.7 eV)显著低于均相催化体系,揭示其高效催化活性。
吸附行为模拟进一步显示,O₂分子在Fe-O终止表面以超氧态(O₂⁻)吸附(吸附能-1.2 eV),并通过差分电荷密度(Δρ)分析证实Fe³⁺→O₂的电子转移机制,激活O-O键以降低解离能垒;而H₂O分子在Fe位点的解离吸附(ΔG=-0.5 eV)生成表面羟基(OH*),为氧化反应提供质子源。
通过对比不同晶面(如{111}与{100})的吸附构型与态密度(DOS)分布,发现{111}面Fe³⁺位点的d带中心位置(-1.8 eV)更接近费米能级,增强反应物轨道杂化效率,定向优化催化路径。
这类计算策略将表面电子结构、吸附动力学与反应能垒定量关联,为设计高活性Fe₃O₄基催化剂提供了理论范式。
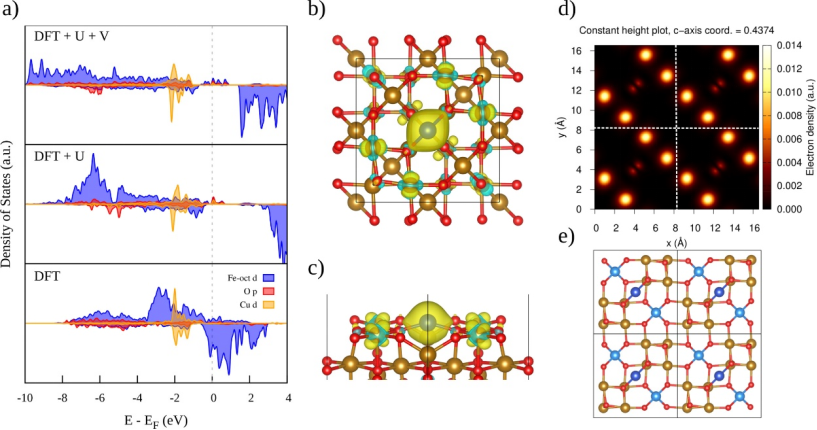
DOI:10.1016/j.rinp.2025.108158
复合材料与界面设计
四氧化三铁(Fe₃O₄)的复合材料设计与界面理论研究通过密度泛函理论(DFT)与分子动力学(MD)揭示了其性能优化机制。
在异质结构体系中,Fe₃O₄与石墨烯或碳基材料的界面电荷分布分析显示,碳基体的π电子与Fe³⁺的d轨道通过界面耦合形成电荷转移通道(差分电荷密度Δρ达0.15 e/ų),显著提升复合材料的导电性(如电导率增加2个数量级)。
同时,Li⁺在Fe₃O₄电极中的嵌入/脱出模拟通过微动弹性带(NEB)方法解析了扩散路径:Li⁺优先沿八面体间隙迁移,扩散能垒低至0.3 eV,但嵌锂过程中晶格膨胀率高达15%(体积膨胀),需通过碳包覆或纳米结构设计缓解应力累积。
进一步结合MD模拟发现,Fe₃O₄/石墨烯界面可抑制锂化过程中的颗粒粉化,界面吸附能(-1.8 eV)稳定电极结构并提升循环寿命。
这类计算从电子耦合与离子输运双维度指导复合材料设计,为高导电、高稳定性的Fe₃O₄基储能与催化材料开发提供了原子级理论支撑。
DOI:10.1016/j.commatsci.2024.113586
经典案例:Fe₃O₄催化反应机理研究

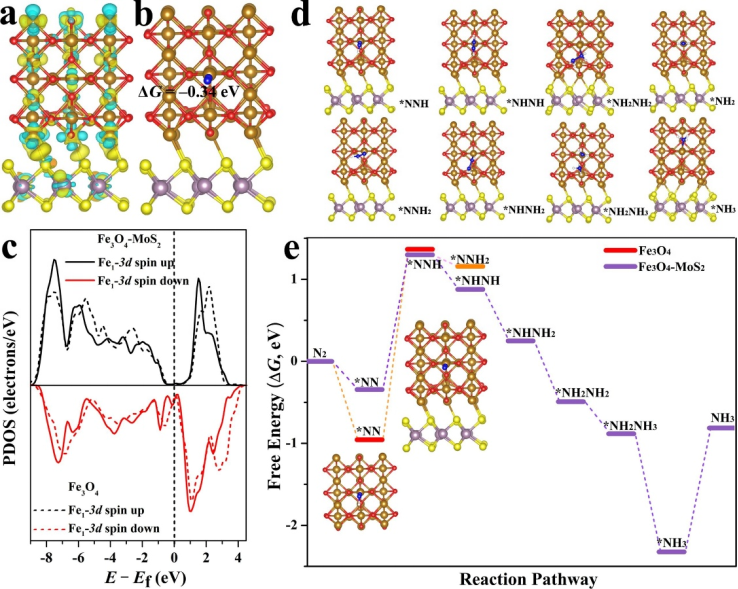
在《Interface engineering ofFe3O4@MoS2 Nanocomposites: High efficiency electrocatalytic synthesis of NH3 under mild conditions》中,研究者借助密度泛函理论(DFT)深入剖析了 Fe₃O₄@MoS₂异质结构在电催化氮还原反应(eNRR)中的作用机制。
计算聚焦于 Fe₃O₄与 MoS₂的界面协同效应,通过构建异质结构模型,揭示了电子转移与催化活性之间的内在联系。
差分电荷密度分析表明,Fe₃O₄向 MoS₂转移了约 0.83e 的负电荷,使 MoS₂表面电荷密度增加,这种电子重排增强了界面处对 N₂分子的吸附能力。
结合部分态密度(PDOS)分析发现,MoS₂的引入调控了 Fe₃O₄中 Fe 原子的 d 轨道分布,使 N₂的吸附自由能(ΔGad)降至 – 0.34 eV,显著提升了反应物的活化效率。
进一步的反应路径模拟显示,N₂分子以端基吸附模式(end-on)锚定在 Fe₃O₄@MoS₂界面的低配位 Fe 原子上,N≡N 键被拉长至 1.130 Å,为后续氢化反应奠定基础。
在对比远端路径(distal)和交替路径(alternating)时发现,交替路径中NNH 中间体的形成能垒更低,且除最终 NH₃脱附步骤外,其余氢化过程均为放热反应,表明该界面可有效降低反应能垒并推动反应正向进行。
与纯 Fe₃O₄(110) 表面相比,Fe₃O₄@MoS₂界面在关键步骤(如N₂到 * NNH 的第一步氢化)的吉布斯自由能变化(ΔG)显著减小,证实了 MoS₂通过电子协同效应优化了 Fe₃O₄的催化活性位点。
DFT 计算还揭示了界面结构对抑制析氢反应(HER)的作用机制:高曲率的 MoS₂纳米片通过 Tolman 效应增强表面张力,减少氢吸附位点,而 Fe₃O₄的多孔结构则提供了高效的质子传输通道,两者协同提升了 eNRR 的选择性。
这些理论发现不仅从原子尺度阐明了 Fe₃O₄@MoS₂的催化本质,也为设计高效过渡金属基电催化剂提供了 “电子结构 – 界面工程” 的双重优化策略,展现了计算化学在指导实验设计中的前瞻性价值。
DOI:10.1016/j.cej.2022.135417
总结
在 Fe₃O₄催化机理研究的基础上,未来研究将沿着多维度理论计算路径展开新探索:通过结合密度泛函理论(DFT)与机器学习的多尺度模拟方法,构建 Fe₃O₄复合材料的性能预测模型,为设计新型催化材料提供高效计算框架;
借助极端条件下的理论模拟,揭示高压 / 高温环境中 Fe₃O₄的相变规律,不仅拓展其在地球深部物质演化等地球科学领域的应用维度,还为极端工况下催化材料的稳定性研究提供依据;同时,利用含时 DFT(TDDFT)技术动态追踪光催化过程中载流子的迁移行为,精准捕捉能量转换的关键节点。
这些研究方向既聚焦于揭示 Fe₃O₄的本征物理化学性质,又紧密对接能源转化、催化反应工程等实际应用场景,通过理论计算与实验设计的深度耦合,为推动铁基功能材料在更复杂体系中的高效利用点亮理论灯塔,让基础研究的 “算力” 切实转化为解决实际问题的 “能力”。
写在最后
#华算科技 #晶体材料 #二维材料 #四氧化三铁 #DFT计算 #磁性分析 #催化机理 #界面效应 #异质结构 #能垒计算 #多尺度模拟