

电子结构分析基于DFT,通过能带结构、态密度(DOS)与差分电荷密度(CDD)揭示材料性能。
COHP量化成键强度,LCAO分析电荷转移,PDOS结合Bader电荷关联活性位点电子特性。
例如,Pt单原子d带中心调控CO吸附能,Cu/GDY体系电荷分布优化催化路径。未来引入机器学习加速筛选,结合DFT+U修正强关联效应,推动单原子催化剂在能源与电子器件中的理性设计,从微观电子行为到宏观性能建立精准模型。



能带结构计算


能带理论基于密度泛函理论(DFT),通过求解单电子薛定谔方程获得周期性体系的电子能级分布,其中局域密度近似(LDA)将交换关联势简化为局域电子密度函数,而广义梯度近似(GGA)引入密度梯度修正以提升计算精度。
其计算流程涵盖结构优化(如VASP中通过ISIF参数调控晶格弛豫)、自洽场迭代求解Kohn-Sham方程获取电荷密度与波函数,以及沿布里渊区高对称路径的能带插值(采用Wannier90方法或线性插值)。
以CdTe/HgTe异质结为例,通过对比直接带隙(CdTe)与间接带隙(HgTe)的能带差异,结合自旋轨道耦合效应计算,揭示了拓扑绝缘体边缘态的电子起源;
而W-N₂O₂体系的能量–波矢(E-k)曲线通过导带狄拉克锥与价带平带特征,阐明其超导与电荷密度波竞争的物理机制。
此类计算以E-k色散图为核心输出,结合态密度投影(PDOS)解析轨道贡献,为二维材料、拓扑物态等前沿领域的电子结构设计提供定量理论框架。

DOI:10.1016/j.chemphys.2022.111674
态密度(DOS)分析
态密度用于描述单位能量区间内电子态的分布情况,主要分为总态密度(TDOS)和投影态密度(PDOS)。
总态密度通过积分所有 k 点的电子态来反映材料整体的电子填充特性,在费米能级处,金属的总态密度非零,而半导体因存在带隙其总态密度在此处为零。
投影态密度则是将总态密度进一步分解为原子轨道(s/p/d/f)的贡献,例如在分析过渡金属的催化活性时,可通过其 d 轨道的投影态密度来研究催化活性中心的 d 带中心位置。
在可视化技术方面,通常借助 VASP+VTST 或 VESTA 等工具生成二维或三维的 DOS 图,以直观呈现态密度分布。如对 Pt1δ+/TiO2 体系的投影态密度分析显示,Pt 的 5d 轨道与 O 的 2p 轨道存在杂化现象,这为金属 – 载体之间的电荷转移机制提供了有力佐证,有助于深入理解材料的电子结构与物理化学性质之间的关系。
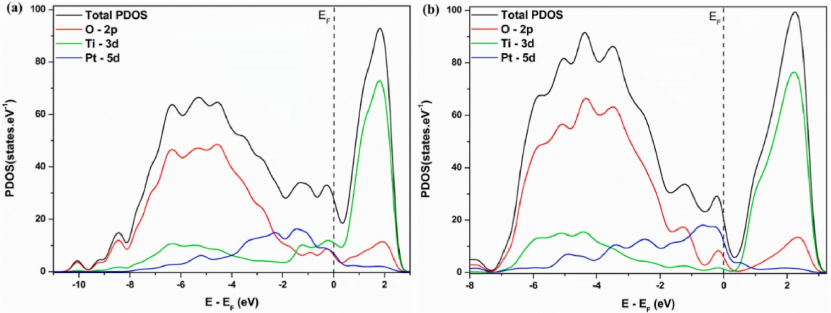
DOI:10.1016/j.physb.2025.417201
局域电荷分析(LCAO)
局域电荷分析(LCAO)基于原子轨道线性组合(LCAO)方法,其数学表达式为 ϕMO=∑iciχiAO,其中 χiAO 代表原子轨道基函数,系数 ci 通过 Hartree-Fock 或 DFT 自洽计算确定,该方法通过将分子轨道表示为原子轨道的线性组合,实现对体系电荷分布与转移特性的精细分析。
在实际应用中,LCAO 方法可有效揭示界面电荷分布与催化活性位点的关联,例如针对 Cu1/GDY 催化剂体系,通过分析 Cu-3d 轨道与 C-2p 轨道在分子轨道中的权重贡献,能够明确苯羟基化反应中起关键作用的活性位点,为催化机理研究提供直接依据。
此外,结合 Bader 电荷分析技术,LCAO 方法还可定量描述单原子与载体间的电荷转移量,如在 Pt1δ+/TiO2 体系中,通过计算得出 Pt1δ+ 携带的正电荷,直接证明其电子向 TiO2 载体流失的过程,这种电荷转移特征与材料的电子结构和化学活性密切相关。
该方法通过数学建模与实际体系的结合,为深入理解原子间电荷相互作用、揭示催化反应活性中心的本质提供了重要的理论与计算工具,在材料科学与催化化学领域具有广泛的应用价值。
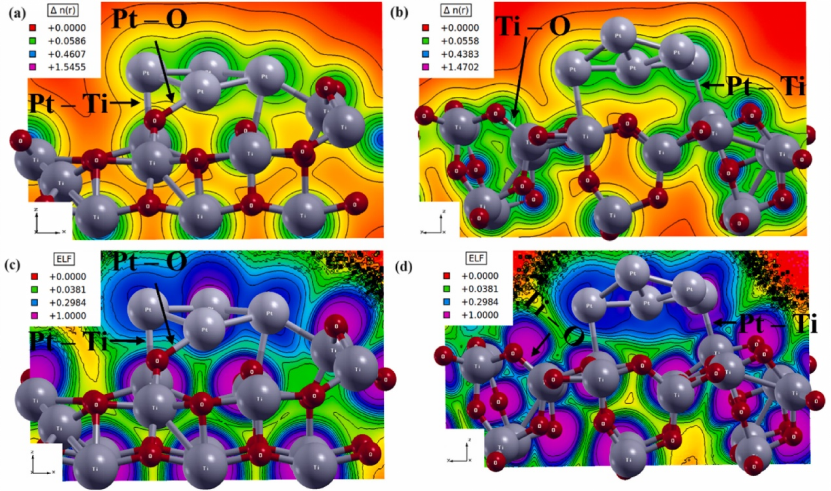
DOI:10.1016/j.physb.2025.417201
分子轨道(MO)对称性与杂化
分子轨道理论以对称性匹配原则为核心,通过原子轨道线性组合(LCAO-MO)方法构建成键轨道与反键轨道,从而揭示分子或材料中电子的运动状态与化学键本质。
其中关键机制包括 σ/π 杂化与轨道能级对齐:在 σ/π 杂化过程中,如 CO 分子吸附于 Fe 单原子催化剂表面时,C 原子的 2p 轨道与 Fe 原子的 3d 轨道通过对称性匹配,形成 σ 供键(电子从 C 向 Fe 转移)与 π 反键(电子从 Fe 反传回 C 的空轨道),这种杂化作用直接影响分子的吸附构型与反应活性;
而轨道能级对齐机制则体现在体系的最高占据分子轨道(HOMO)与最低未占据分子轨道(LUMO)的能级差对反应路径的调控上,例如 W-N2O2 活性中心的 LUMO-HOMO 能级差可决定氮还原反应(NRR)中不同中间体的吸附能垒,进而影响反应路径的选择性。
在可视化分析中,借助 Multiwfn 或 Gaussian 等计算工具生成的轨道等值面图,能够直观呈现分子轨道的空间分布特征,如苯酚产物的前线分子轨道(HOMO/LUMO)分布可清晰显示电子云在分子骨架上的离域情况,为理解化学反应中轨道重叠、电荷转移及活性位点的作用提供可视化依据。
该理论通过将量子力学原理与轨道相互作用机制相结合,不仅阐明了化学键的本质,更在催化反应机理、材料电子结构分析等领域发挥重要作用,成为连接微观轨道特性与宏观化学性质的关键理论工具。
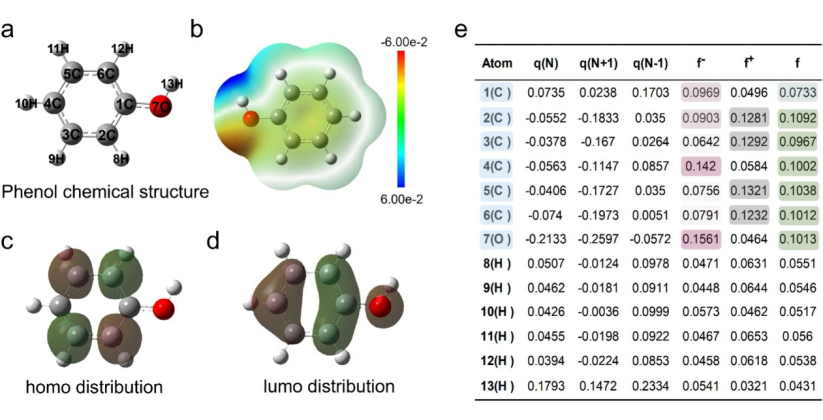
DOI:10.1016/j.jcis.2025.137391
差分电荷密度(CDD)
差分电荷密度(CDD)通过计算式 Δρ=ρtotal−∑iρatomi 表征体系中界面电荷的重排情况,其中 ρtotal 为体系总电荷密度,∑iρatomi 为各孤立原子电荷密度的叠加,二者的差值直观反映了原子间相互作用导致的电荷重新分布。
在吸附体系研究中,以 W-NO/NC 体系为例,其 CDD 图显示 NO 分子吸附后,W 原子周围出现明显的电子累积区域(黄色区域),这一特征表明 W 与 NO 分子之间存在强电子相互作用,电荷在界面处发生显著重排,进而影响吸附构型与反应活性。
而在异质结界面分析中,当石墨烯负载 Pt 单原子时,CDD 图清晰揭示界面处电子从 Pt 单原子向 C 基底转移的过程,这种电荷转移行为与材料的电子结构密切相关,直接影响异质结的界面性质与催化性能。
作为一种有效的电荷分析工具,CDD 通过量化电荷密度的差异,将原子间的相互作用转化为可视化的电荷分布特征,不仅能够直观展现界面处电子的聚集或流失情况,还为深入理解材料界面的电子转移机制、吸附行为及化学键本质提供了重要依据,在催化材料设计、异质结性能优化等领域具有关键的应用价值,帮助研究者从电荷重排的角度剖析微观界面过程与宏观材料性质之间的内在联系。
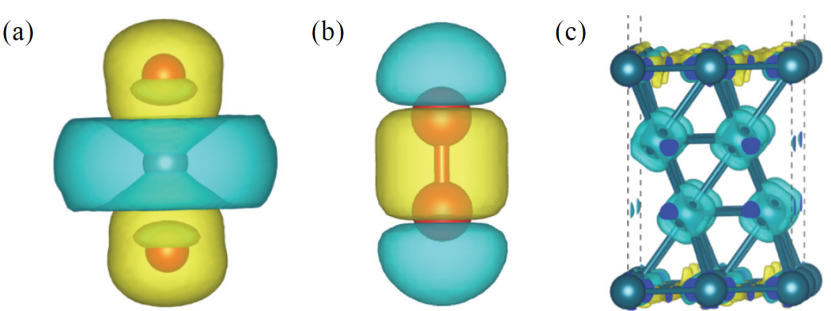
DOI:10.3866/PKU.DXHX202107125
晶体轨道哈密顿布居(COHP)
晶体轨道哈密顿布居(COHP)是一种用于量化原子间成键强度的重要方法,其通过分析晶体轨道中电子分布对成键的贡献,以数值正负表征键的性质 —— 负值表示成键态(电子分布利于化学键形成),正值对应反键态(电子分布削弱化学键),而积分 COHP(ICOHP)则可定量评估键能大小,为揭示原子间相互作用强度提供直接依据。
在金属 – 载体相互作用研究中,以 Rh/ZrO₂与 Fe/ZrO₂体系为例,Rh-O 键的 ICOHP 值为 – 3.28 eV,显著强于 Fe-O 键的 – 1.91 eV,这一差异直接解释了 Rh/ZrO₂在氧析出反应(OER)中表现出更高活性的原因,表明更强的金属 – 载体成键作用可有效优化反应中间体的吸附能垒。
而在催化活性调控领域,Co₂N₅模型中 Co-O 键的 ICOHP 值为 – 0.47 eV,这一适度的成键强度恰好平衡了反应过程中吸附与脱附的能垒,使得该体系在氧还原反应(ORR)与氧析出反应(OER)中均展现出优异的动力学性能,体现了COHP 方法在指导催化剂设计时通过调节原子间成键强度优化催化活性的关键作用。
COHP 通过将量子化学计算与键能定量分析相结合,不仅为阐明材料中原子间相互作用的本质提供了微观视角,更在催化反应机理解析、高性能催化剂开发等领域发挥重要作用,成为连接电子结构与宏观催化性能的关键桥梁。
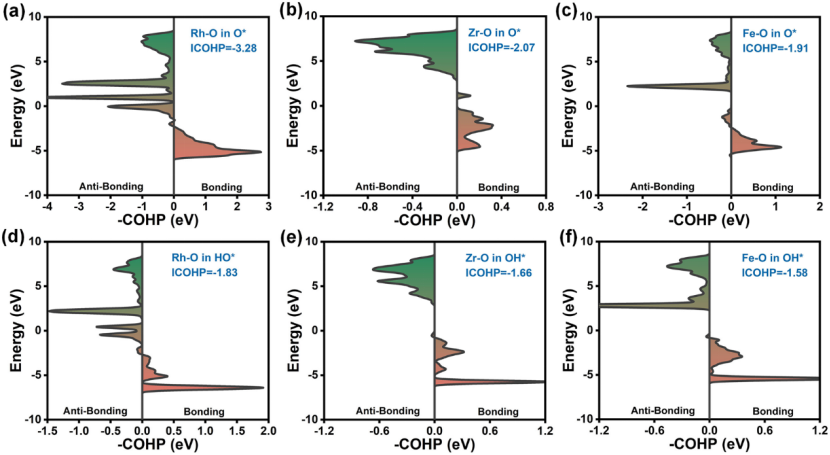
DOI:10.1007/s40820-024-01403-7
态密度投影与电荷关联性
态密度投影(PDOS)与 Bader 电荷分析相结合,能够建立材料电子结构特征与宏观性能之间的内在关联,为揭示催化活性、吸附行为等物理化学性质的本质提供关键依据。
基于 d 带中心理论,过渡金属的 d 带中心(εd)位置是衡量其与吸附物结合能力的重要指标,当 εd 上移时,金属与吸附物的轨道杂化作用增强,进而提升结合能,例如 Pt (111) 表面的 d 带中心与 CO 吸附能呈现显著的线性关系,验证了该理论在预测吸附行为中的有效性。
而在电荷再分布分析中,通过 PDOS 对原子轨道贡献的分解与 Bader 电荷对原子电荷得失的量化,可共同揭示活性位点的电子结构特征。
如 Cu1/GDY 催化剂中,Cu 单原子的 3d 轨道 PDOS 显示特定能量区间的态密度分布,结合其 Bader 电荷为 + 0.32e 的结果,直接证实该位点因电子流失而呈现 Lewis 酸性,这种酸性位点对反应物的活化及催化反应路径的调控起到关键作用。
二者的结合不仅从轨道贡献与电荷转移两个维度刻画了电子结构的细节,更通过具体案例展现了如何从微观电子分布特征推导材料的化学性质与催化性能,为设计具有特定电子结构的功能材料提供了理论指导,成为连接量子化学计算与实际应用的重要分析手段。
DOI:10.1073/pnas.1006652108
结论
电子结构分析框架借助多尺度计算手段,从能带结构解析、态密度表征到成键特性分析等多个维度,系统且全面地揭示了原子尺度下的电子行为规律,为理解单原子材料的物理化学性质与催化机制提供了微观视角的理论支撑。
随着计算材料学的发展,该框架的未来研究将聚焦于三大方向:一是引入机器学习技术加速高通量计算筛选,通过数据驱动优化单原子催化剂的设计效率;
二是开展非平衡态电子输运过程的模拟研究,以更贴近实际反应条件下的电子动态行为分析;三是针对强关联体系进行 DFT+U 方法的精确修正,突破传统密度泛函理论在处理电子强关联效应时的局限性。
这些发展方向不仅有望深化对单原子电子结构的认知,更将推动该分析框架在能源催化、电子器件等领域的实际应用,为开发高性能功能材料提供更精准的理论指导。