

说明:Lewis酸位点是电子对受体,DFT可通过电子结构、电荷密度、吸附模型等分析其特征与强度。研究聚焦MOFs、金属氧化物、沸石中位点调控,如Zr-MOF缺陷增强酸性,Sn-BEA沸石催化乳酸脱水。DFT为催化剂设计提供理论支撑,未来需发展高精度泛函与机器学习。
什么是Lewis酸位点?
Lewis酸位点(Lewis Acid Sites, LAS)的本质是电子对受体,由G. N. Lewis于1923年提出,根据IUPAC定义,其为能接受电子对并通过与Lewis碱形成配位共价键生成加合物的分子实体。
在密度泛函理论(DFT)计算中,Lewis酸位点的特征可通过电子结构分析精准识别,具体表现为:金属中心的不饱和位点,如金属有机框架(MOF)中的开放金属位点(OMS)或沸石中Sn-BEA等骨架金属取代位点,其空轨道可作为电子对接受体;表面缺陷或空位,典型如CeO₂、TiO₂等金属氧化物的氧空位,通过局部电子结构重构增强Lewis酸性;正电荷富集区域,通过电荷密度分析定位Bi³⁺、Hf⁴⁺等阳离子周围的局部正电势区域,该类区域因电荷失衡呈现电子对接受能力。
DFT通过量化分析轨道能级、电荷分布及缺陷形成能,为Lewis酸位点的精准识别与酸性强度调控提供了原子尺度的理论支撑。


DOI:10.1002/cctc.202300471
DFT计算中Lewis酸位点
电子结构分析
在密度泛函理论(DFT)计算中,Lewis酸位点的电子结构分析通过量化电子分布与能级特征实现对Lewis酸性的精准表征。
HOMO-LUMO能隙(ΔE)是关键指标,其中最低未占分子轨道能量直接反映位点接受电子对的能力——越低(即轨道能量越接近费米能级),接受电子对的能力越强。
ΔE越小,表明电子从最高占据轨道(HOMO)跃迁到LUMO所需能量越低,Lewis酸性越强。需注意,传统DFT泛函(如PBE)存在带隙低估问题,可能导致计算偏差,需结合ΔSCF方法(自洽场能量差法)校正以提升精度。
轨道成分分析通过投影态密度(PDOS)识别LUMO轨道的原子及轨道贡献,例如TiO₂(001) 表面的LUMO若由Ti³⁺的3d空轨道主导,表明该位点因存在未占据的d轨道而具备电子对接受能力,可明确其Lewis酸位点属性。
DFT通过整合HOMO-LUMO能隙量化与轨道成分解析,从分子轨道能级与电子分布角度构建了Lewis酸性的理论描述框架,为催化剂活性位点筛选及酸性调控提供了原子尺度的理论依据。
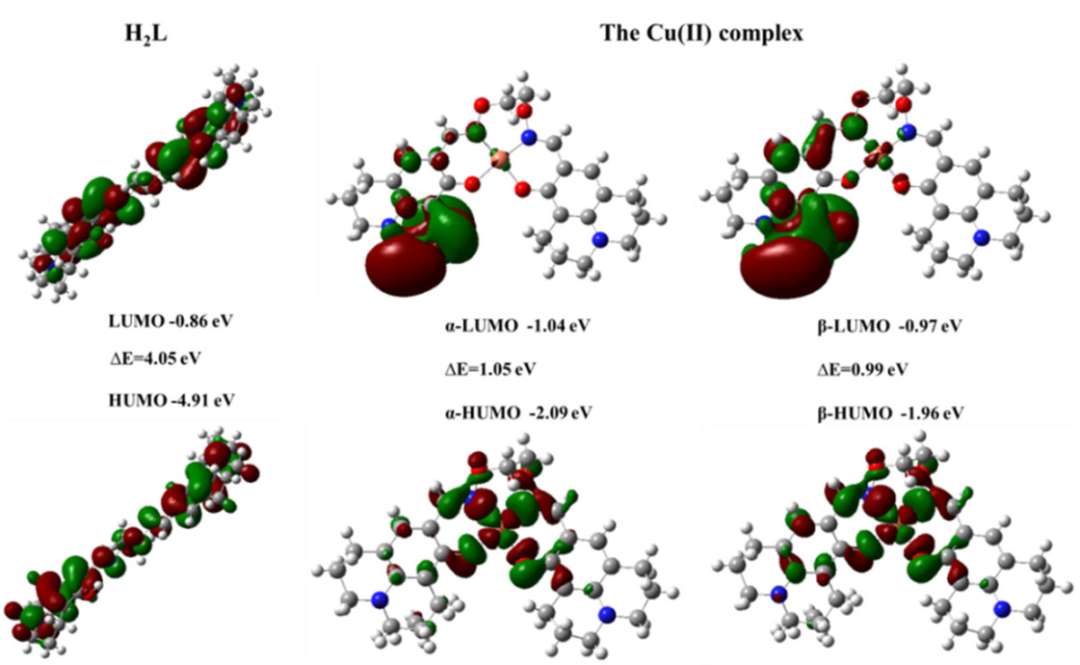
DOI:10.1080/00958972.2022.2123738
电荷密度与静电势分析
在密度泛函理论(DFT)计算中,电荷密度与静电势分析是表征Lewis酸位点(LAS)的关键手段。
通过Bader电荷划分可量化金属中心的局部电荷分布,揭示Lewis酸性的起源与强度——如SO₄²⁻修饰的CeO₂体系中,Ce⁴⁺的Bader电荷从+1.75e增至+1.92e,电荷正移使其对电子对的吸引能力增强,直接体现Lewis酸性的提升。
静电势分布图则通过可视化表面电势场,直观定位Lewis酸位点:在WO₃表面,W原子因低电子密度形成正电势区域(常以蓝色标记),该区域作为电子对受体可高效吸附烃类分子的孤对电子,其作用机制可通过图19所示的W原子电荷重分布过程进一步佐证 —— 电荷从周边氧原子向W中心迁移,导致局部正电势累积,强化了Lewis酸位点的电子接受能力。
DFT结合Bader电荷分析与静电势mapping,从电荷量化与空间分布双维度构建了Lewis酸位点的精准表征框架,为催化材料的酸性调控提供了原子尺度的理论依据。
吸附模型与结合能计算
在密度泛函理论(DFT)计算中,Lewis酸位点的酸性强度可通过探针分子(如NH₃、CO)的吸附模型与结合能分析精准量化。
吸附能作为核心指标,其负值大小直接反映位点与探针分子的结合能力——负值越大,Lewis酸性越强:例如NH₃在SO₄²⁻修饰的CeO₂(111) 表面吸附能为-0.96 eV,较未修饰体系的-0.40 eV显著增强,印证了表面硫酸根修饰对Lewis酸性的提升作用;CO在TiO₂(001) 表面以单齿碳酸盐形式吸附时吸附能达-0.85 eV,而在 (101) 面仅呈现-0.15 eV的弱吸附,揭示了晶面差异对酸性位点活性的影响。
振动频率分析则通过探针分子特征峰的位移量建立酸性关联,以CO为例,其C≡O伸缩振动的红移量(Δν)与Lewis酸性强度呈正相关,该分析需结合DFT的频率计算模块,通过量化振动模式变化实现酸性的间接表征。
DFT借助吸附能与振动光谱的联合分析,为Lewis酸位点的酸性强度排序及催化材料设计提供了可量化的理论依据。
DOI:10.1021/jp507443k
DFT研究Lewis酸位点的核心进展
MOFs中的可调控Lewis酸位
在金属 – 有机框架(MOFs)的Lewis酸位点调控研究中,密度泛函理论(DFT)已成为揭示位点活性与酸性调控机制的核心工具。
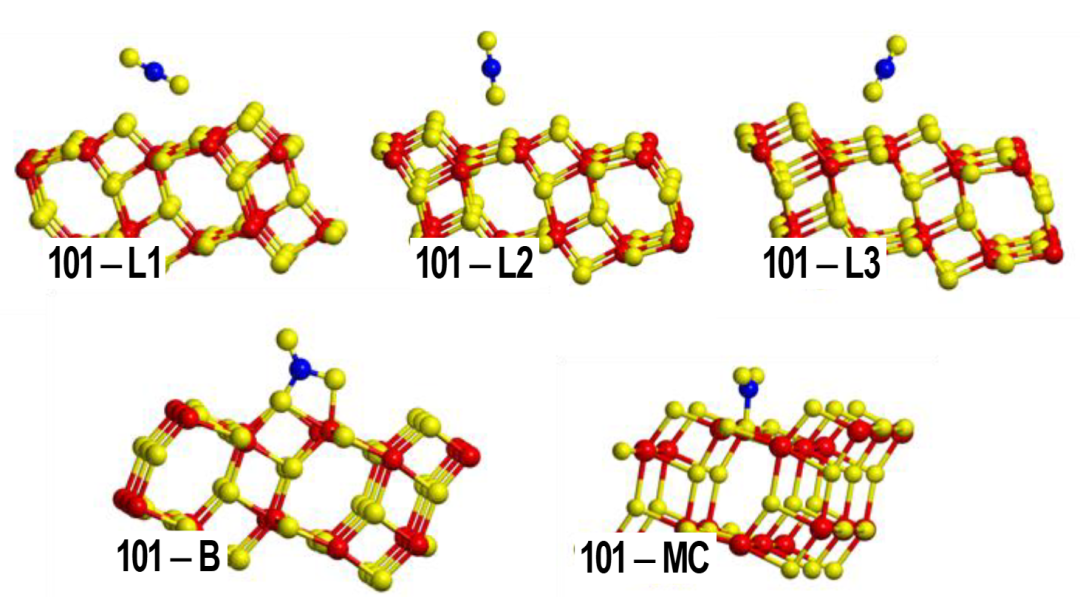
以Hf-MOF-808为例,通过溶剂热合成策略可精准调控材料中Lewis酸与Brønsted酸的比例,DFT计算表明,水热合成条件能显著增加Hf⁴⁺位点的配位不饱和度,使其最低未占分子轨道(LUMO)能级下降0.8 eV,从而增强对电子对的接受能力,直接体现为Lewis酸性的提升。
另一典型体系SU-101 MOF中,骨架内Bi³⁺开放位点作为高效Lewis酸中心,可催化环氧化物开环反应,DFT计算进一步揭示其对甲醇的吸附能垒仅为0.3 eV,显著低于乙醇的0.5 eV,这一能量差异直接解释了实验中甲醇参与反应时转化率达99.8%(乙醇体系为96.8%)的现象。
DFT通过量化配位环境对金属中心电子结构的影响(如LUMO 能级、吸附能垒),不仅阐明了MOFs中Lewis酸位点的调控机制,更为设计高活性催化位点提供了从原子尺度到宏观性能的理论桥梁。
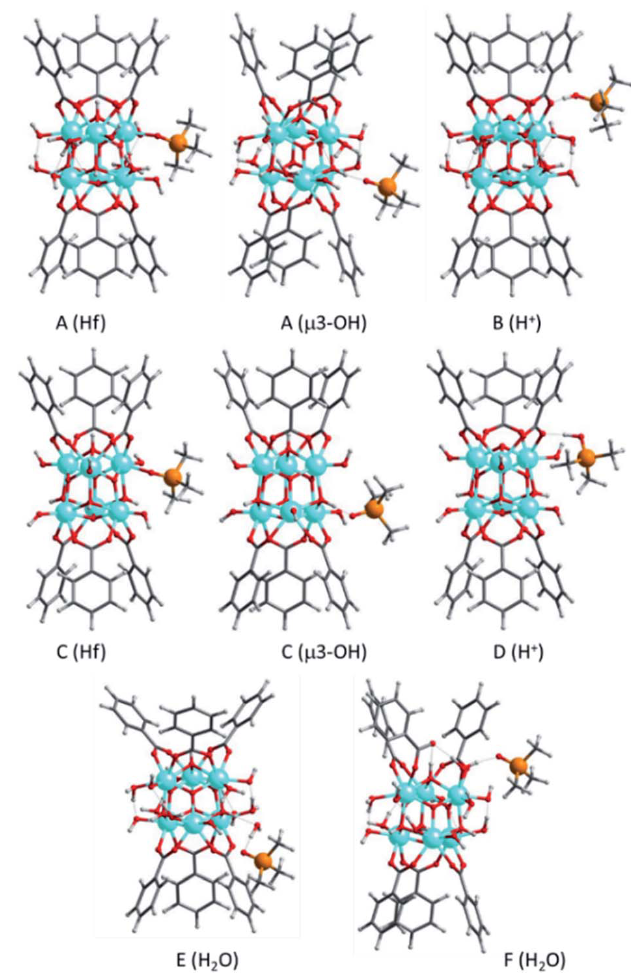
DOI:10.1039/d1sc02833b
金属氧化物表面缺陷工程
在金属氧化物表面缺陷工程领域,密度泛函理论(DFT)为揭示Lewis酸位点的构效关系提供了关键理论支撑。
以CeO₂氧空位(Oᵥ)为例,DFT计算表明,氧空位的存在导致周边Ce³⁺位点的最低未占分子轨道(LUMO)能级下降1.2 eV,显著增强其电子对接受能力,使NH₃吸附能达−0.96 eV,较完美表面提升约40%,从而加速选择性催化还原(SCR)反应的速率控制步骤,将反应能垒降至15.68 kcal/mol。
另一典型体系Cs₃Bi₂Br₉钙钛矿中,表面暴露的Bi³⁺位点通过LUMO轨道(主要由Bi-6p轨道贡献)接受环氧乙烷分子的孤对电子,DFT计算显示该活化过程的能垒较无缺陷表面降低0.7 eV,根源在于Bi³⁺位点的局部正电荷富集与空轨道配位能力的协同作用。
DFT通过量化缺陷诱导的电子结构变化(如LUMO能级位移、吸附能差异),精准阐释了金属氧化物表面缺陷如何调控Lewis酸性:氧空位通过价态转变(Ce⁴⁺→Ce³⁺)重构局部电子环境,而钙钛矿表面低配位Bi³⁺位点则通过轨道杂化增强电子接受能力。
这些研究不仅揭示了缺陷工程提升催化活性的微观机制,更为设计高活性Lewis酸位点提供了 “缺陷 – 电子结构 – 催化性能” 的跨尺度理论框架,推动了金属氧化物在环境催化、有机合成等领域的应用优化。
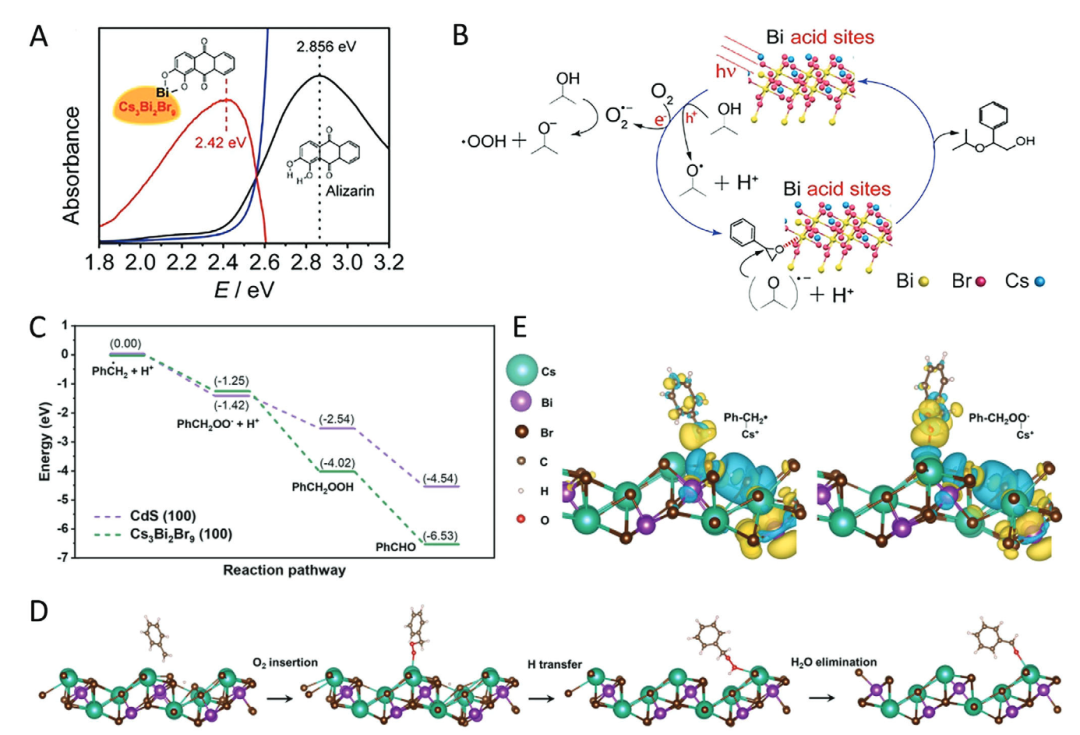
DOI:10.1002/advs.202402471
沸石中的骨架取代位点
在沸石催化材料研究中,骨架金属取代位点作为Lewis酸活性中心的构效关系可通过密度泛函理论(DFT)精准解析,以Sn-BEA沸石为例,周期DFT计算揭示了Sn⁴⁺取代骨架Si原子后引发的电子结构重构及其催化机制。
当Sn⁴⁺进入BEA沸石骨架时,其较低的电负性与配位几何结构导致局部电子环境改变,最低未占分子轨道(LUMO)能级降至-3.5 eV,显著增强了位点对电子对的接受能力。
在乳酸脱水反应中,该Sn⁴⁺位点通过空的5s/5p轨道吸附乳酸分子,形成五配位中间体,其中Sn⁴⁺与乳酸的羟基氧原子形成配位共价键,使C-OH键活化。
DFT计算显示,该中间体的脱水反应能垒仅为0.8 eV,远低于纯硅BEA沸石的无催化路径,根源在于Sn⁴⁺位点的Lewis酸性促进了羟基氢的解离与碳氧键的断裂。
这种骨架取代诱导的LUMO能级调控与配位中间体形成机制,不仅阐明了Sn-BEA沸石高催化活性的微观起源,更建立了 “金属取代 – 电子结构 – 反应路径” 的理论关联,为设计高选择性沸石基Lewis酸催化剂提供了原子尺度的调控策略,推动了其在生物质转化、精细化学品合成等领域的应用研究。
DOI:10.5267/j.ccl.2019.5.002
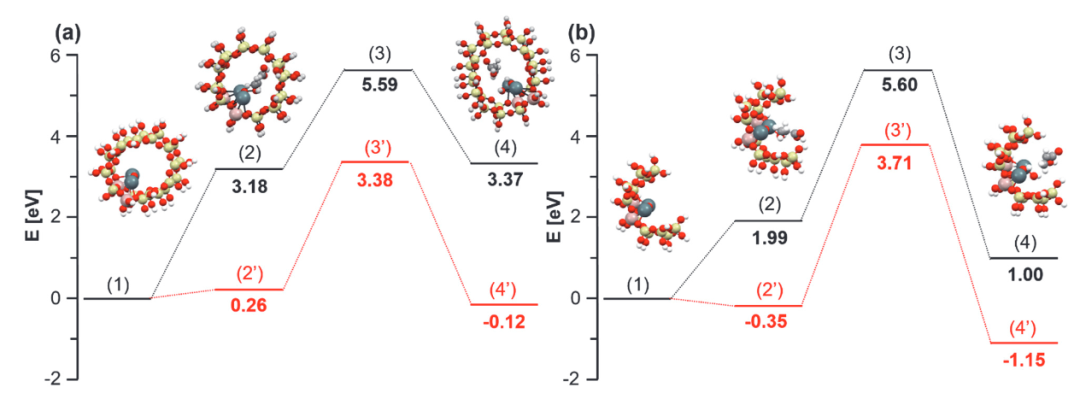
MOF缺陷增强Lewis酸性的应用
在2024年《Nature Catalysis》的一项研究中,通过密度泛函理论(DFT)系统揭示了Zr-MOF缺陷模型中Lewis酸性增强的微观机制。研究构建了移除配体形成不饱和Zr⁴⁺位点的缺陷模型,通过电子结构分析、探针分子吸附及反应能垒计算等多尺度模拟,阐明了缺陷诱导的电子结构重构与催化性能提升的关联。
首先,电子结构分析显示,缺陷位点的Zr⁴⁺最低未占分子轨道(LUMO)能级从-2.1 eV显著降至-3.0 eV,反映出缺陷导致的电子接受能力增强。
Bader电荷分析进一步量化了电荷重分布:缺陷位点Zr⁴⁺的电荷从+1.82e增至+2.15e,表明配体移除后Zr⁴⁺周围电子密度降低,局部正电荷富集,强化了Lewis酸位点的本质特征。
探针分子吸附实验验证了Lewis酸性的提升:NH₃在缺陷表面的吸附能为-0.46 eV,显著低于完美表面的-0.25 eV,表明缺陷位点对电子对的捕获能力更强。
电荷差分密度图(CDD)直观显示,吸附过程中电子从NH₃的孤对电子轨道向Zr⁴⁺的空轨道转移,蓝色区域(电子富集区)集中于Zr⁴⁺周围,明确了电子转移路径与配位作用机制。
在DCPD氢化反应中,缺陷位点的催化活性提升体现在反应能垒的显著降低:决速步能垒从完美表面的1.15 eV 降至0.72 eV,根源在于缺陷诱导的LUMO能级下降与电荷重分布协同优化了反应路径。
核心图示对比了两种表面的反应路径能垒,并揭示了过渡态结构中Zr⁴⁺与H₂键长的动态变化 —— 缺陷位点的Zr-H键长缩短0.12 Å,表明H₂分子在缺陷位点更易解离活化,从而降低反应能垒。
该研究通过DFT计算建立了 “缺陷 – 电子结构 – 催化性能” 的理论框架:配体移除导致Zr⁴⁺配位不饱和,引发LUMO能级下移与电荷密度重新分布,两者共同增强Lewis酸性,进而提升吸附能力与反应动力学。
这一机制不仅为MOF缺陷工程提供了理论依据,也为设计高效Lewis酸催化剂提供了 “电子结构精准调控” 的新思路,推动了MOF在催化、能源等领域的应用优化。
DOI:10.1016/j.jcat.2024.115747
总结
DFT理论计算通过电子结构分析(LUMO能级、电荷分布)、吸附模型(结合能、振动频率)及反应路径模拟(过渡态能垒)多维度量化Lewis酸位点性质。
近五年研究聚焦缺陷工程(氧空位、MOF不饱和位点)和杂原子掺杂(Sn/Bi取代)对Lewis酸性的调控机制,为理性设计催化剂提供理论基石。
未来需发展高精度泛函(如SCAN-rVV10)及机器学习加速的活性描述符挖掘(如LUMO能级与吸附能的构效关系)。