

说明:DFT框架下的化学键分析通过多维度方法展开,轨道相互作用分析借助NBO、COHP揭示键本质,电子密度拓扑分析利用QTAIM、ELF定位键特征,电荷与键级量化及能量分解深化理解。
Ta₄B₁₈案例中,多种方法证实其通过多中心键稳定并具球芳香性,机器学习正推动键分析高效化。
DFT化学键分析


在DFT框架下进行化学键分析时,可通过多维度计算方法揭示原子间相互作用的本质。
轨道相互作用分析中,自然键轨道(NBO)理论将离域分子轨道转化为定域化化学键图像,通过量化键级、供体–受体相互作用能(E (2))及轨道占据数,清晰呈现键的极性、杂化程度与超共轭效应,如在卤代环丙烷研究中,NBO二阶微扰能证实C-X 键的σ→σ*超共轭强度按F>Cl>Br依次减弱,其轨道等值面图还可直观展示电子离域路径。
而晶体轨道哈密顿布居(COHP)专为周期性体系设计,通过LOBSTER输出的COHP数据,精准量化特定原子对在不同能量下的成键/反键作用。
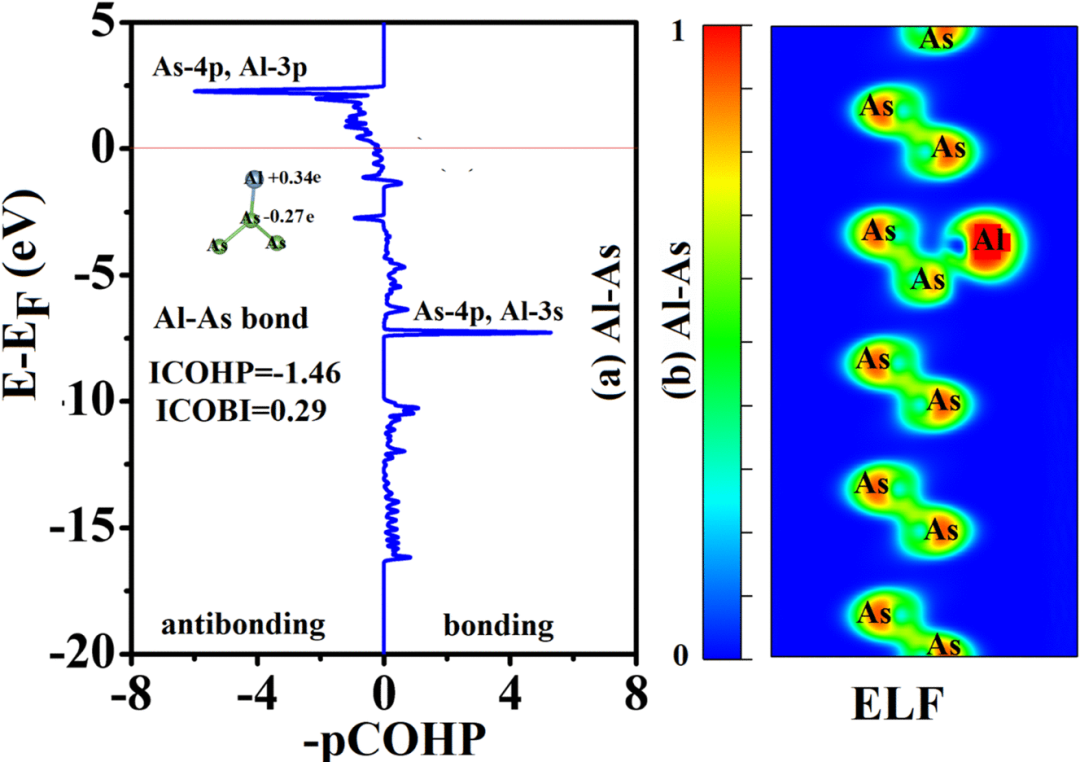
DOI:10.1039/d5ra00232j
电子密度拓扑分析借助量子化学拓扑(QTAIM)与电子定域化函数(ELF)实现深度解析。
基于Bader理论的QTAIM通过电子密度ρ(r) 的拉普拉斯∇²ρ定位键临界点(BCP),当ρ(BCP)>0.3 a.u.且∇²ρ(BCP)时为共价键,如NaCl中∇²ρ=+0.24对应离子键,该方法成功预测西格列汀分子内H…N氢键的ρ=0.028 a.u. 及≈5 kcal/mol键能。
ELF则通过揭示σ/π键与孤对电子的空间分布,呈现不同化学键的电子定域特征,例如苯分子中轮胎状π 电子盆(η≈0.8)与乙烷σ键的纺锤形盆形成鲜明对比。
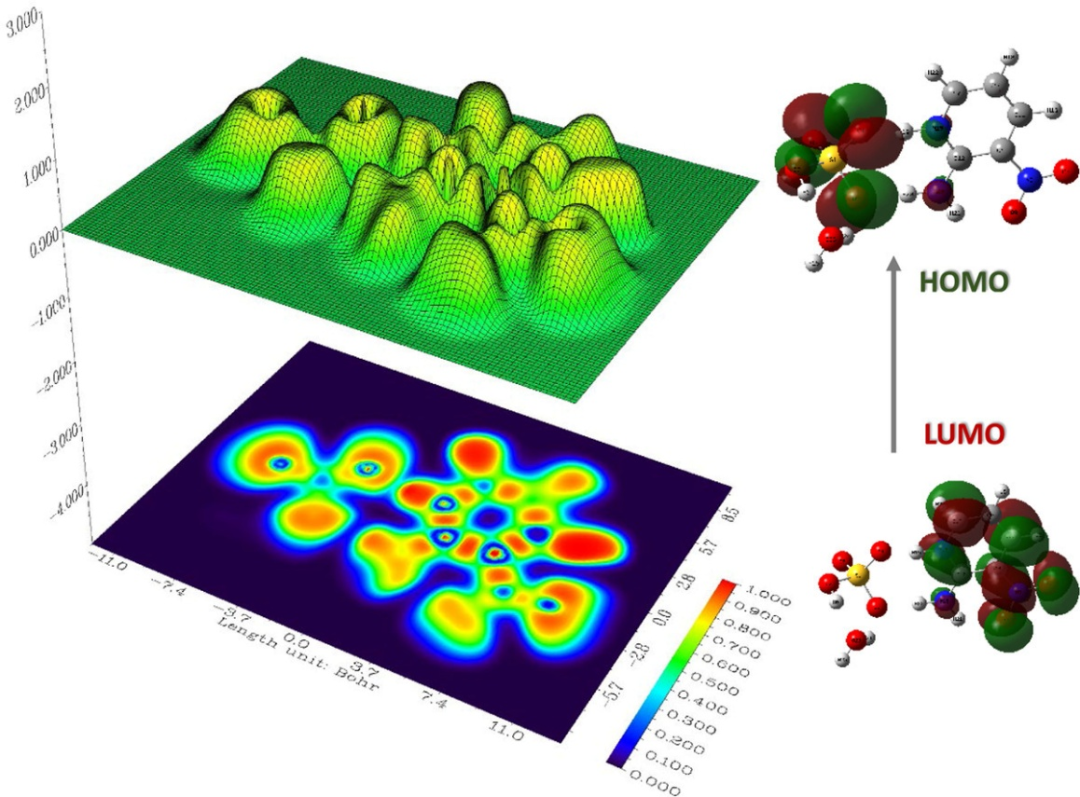
DOI:10.1016/j.molstruc.2025.141839
电荷与键级量化方法各有侧重:Hirshfeld电荷收敛快,适合有机分子的反应活性位点预测,但对基组敏感;ADCH电荷包含偶极校正,适用于极性溶剂环境却计算成本较高;Wiberg键级直观体现共价键强度,在共轭体系键序分析中优势显著,却不适用于金属键体系。
能量分解分析(EDA)则将键合能ΔE解耦为静电作用、泡利排斥与轨道作用,深化对复杂化学键的理解。
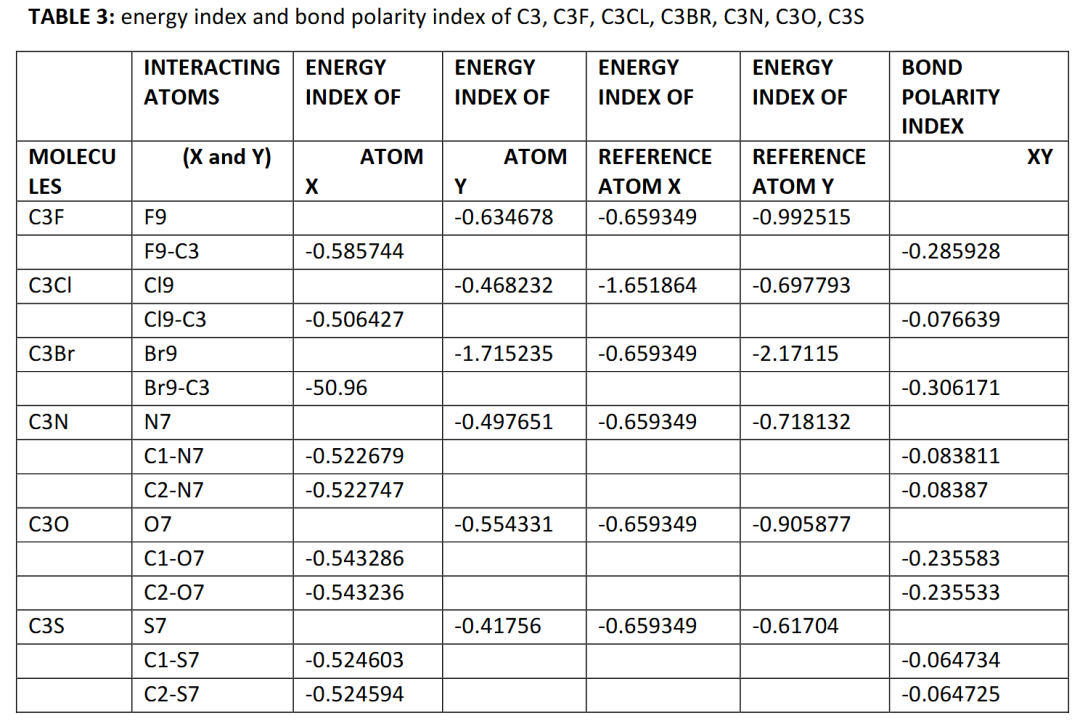
DOI:10.21203/rs.3.rs-136338/v1
近年来,机器学习与键分析的结合推动高通量筛选发展,通过120,000+DFT计算训练的神经网络,可将键能预测误差控制在,大幅提升新材料研发效率。
这些方法共同构建了从轨道相互作用到电子密度分布、从电荷转移到能量分解的完整分析体系,使科研人员能在原子尺度解码化学键的本质,为催化机制、材料设计等领域提供关键理论支撑。
化学键分析的应用
Zhang Y.等人于2021年在《Journal of Physical Chemistry Letters》发表的研究,对新型金属硼球烯Ta₄B₁₈开展了系统的化学键分析。
研究伊始,在PBE0/def2-TZVP计算级别下对Ta₄B₁₈进行几何优化,确定其具有完美四面体结构,呈现D₂d对称性,为后续深入探究其化学键本质奠定基础。
在化学键分析环节,研究采用多种方法交叉验证。运用AdNDP化学键分析发现,Ta₄B₁₈中存在4个符合Hückel规则的6c-2e键,以紫色标注呈现,该结果证实了其具备球芳香性特征;ELF等值面分析则从电子定域化角度,通过三维等值面展示出Ta-B键存在局部化电子盆,电子定域化函数值η达0.75,直观地证实Ta-B键具有强共价性。
而COHP曲线分析进一步量化成键强度,Ta-B键在费米能级下的成键态积分ICOHP为– 5.2 eV,显著强于Ta-Ta键的– 3.8 eV ,表明Ta-B键在结构稳定中起到关键作用。
综合多种分析方法的协同论证,研究明确了Ta₄B₁₈通过σ-π-δ多中心键实现结构稳定,这一结论突破了传统金属团簇的成键认知,为金属硼化物的化学键理论研究提供了全新视角,也为后续新型金属团簇材料的设计与开发,在理解成键机制、预测材料性质等方面提供了重要的理论依据和研究范例。
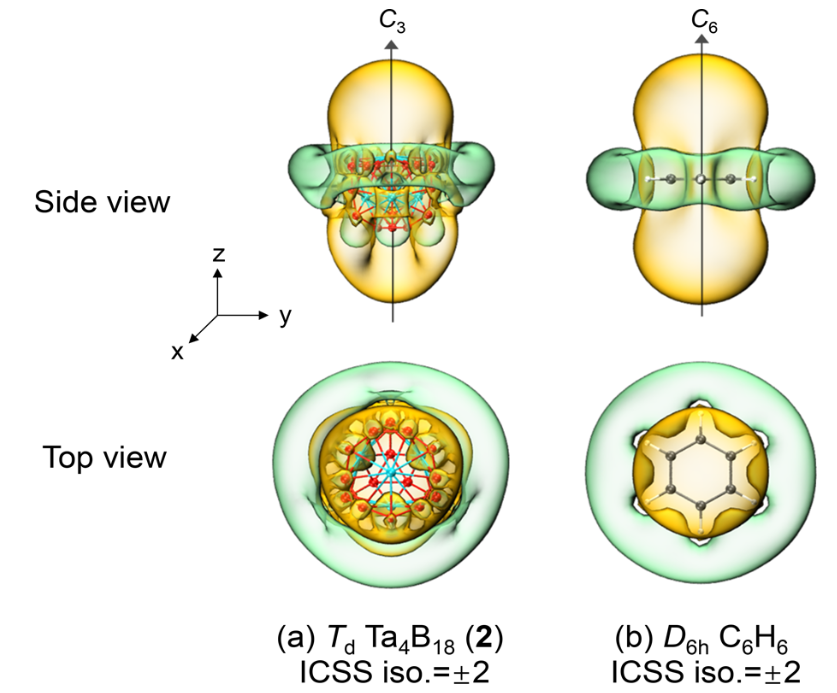
DOI:10.1021/acsomega.1c00828
总结
掌握DFT化学键分析方法相当于拥有探索分子世界的精密工具,多种分析手段从不同维度揭示化学键本质:NBO分析将离域轨道转化为定域化图像,量化轨道相互作用与超共轭效应;QTAIM通过电子密度拓扑定位键临界点,区分共价键与离子键特征;COHP则为周期性体系中的成键/反键作用提供量化依据,不同方法联用可系统解码化学键的量子机制。
随着技术发展,机器学习与量子计算的融合(如NVIDIA量子云平台)正推动键级预测向实时化迈进,未来5年这一趋势将彻底变革材料设计范式,使科研人员能更高效地从原子尺度理解成键规律,为新型功能材料的精准设计与性能优化提供强有力的理论支撑和技术手段。