

Q1:朱老师,教程里有finite temperature DFT的教程吗?想计算温度对于能带的影响
A:算不了温度对电子结构影响
Q2:朱老师,这个报错是什么原因呢?
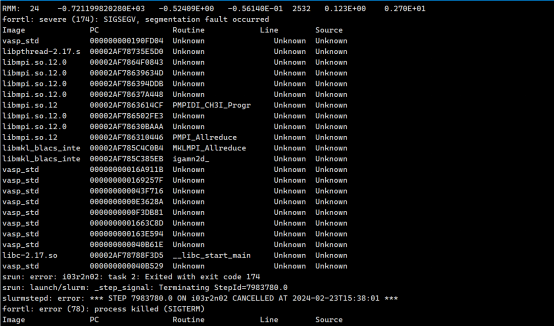

A:设置amix=0.1试试
Q3:朱老师,NiOOH@CoP这个体系构建了一个异质结,研究OER电催化,大概196个原子,用28个核计算,固定了NiOOH底层,截断能450eV,受力收敛为-0.05,电子步收敛为10-4,kpoint为221,看了一下计算时间8000多才算一个离子步,总觉得计算很慢,请问有什么办法提高计算速度?
A:这个没有好办法,可以增加资源
Q4:朱老师,我想问下外加磁场对电催化的影响,要如何去计算?
A:磁场应该不行
Q5:朱老师,为什么我计算出来的dos峰这么尖锐,都看不到细节
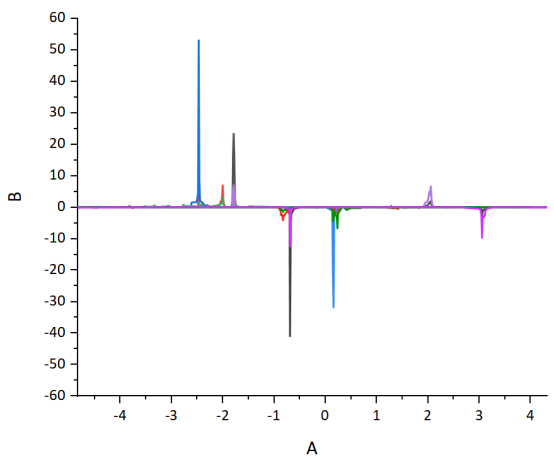
A:ismear用0,sigma用0.1试试
Q6:朱老师,碱性条件下,LOM机制,中间体吸附在金属位点,我列出的步骤是否正确,哪一步有问题
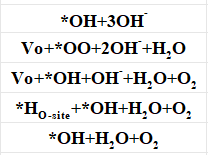
A:最后一步的吸附oh改成自由态水,前面还有一步4oh+*
Q7:朱老师,我在优化晶胞参数的时候用了2*2*2的超胞,此时a b c变二倍了,请问这种情况k点怎么取合适呢?
A:k高到一定程度,能量和晶格就不变了,可以测试一下
Q8:朱老师,请教下静态自洽算电荷K点比优化时要设得大一倍吗,还是可以保持一样?K点的取值是不是根据ka*晶胞a=kb*晶胞b=kc*晶胞c=20-30左右?
A:优化可以靠近20,自洽计算靠近30,条件允许再高些也可以
Q9:朱老师,文献有说表面低配位晶格氧有强碱性,请教下怎么计算低配位晶格氧有哪些?通过bader电荷看吗?
A:碱性的话软件算不了
Q10:朱老师,怎么看晶体晶格氧配位饱不饱和呢?
A:氧没有饱和一说,只有配位数高低
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!