

Q1:朱老师,我想问一下要比较表面slab模型在有无氧空位情况下对HER和OER过程产生的影响。可以把没有氧空位情况下优化好的吸附结构拿来,去掉slab中的一个氧,然后用这个结构直接优化吗?
A:可以的
Q2:朱老师,想问一下催化剂的D带中心越靠近费米能级吸附能力越强,那就代表着催化剂的催化活性越好吗?
A:不是,d带越高只是吸附越强。和催化性能没有关系
Q3:朱老师,吸附能力越强,会导致对中间体较难解吸,从而使得催化活性减弱呢,老师,那这种情况是可能存在的吗?
A:会的,常见
Q4:朱老师,我算出来的材料的静电势是这样的,不知道为什么会这样
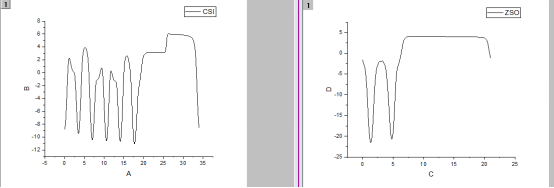

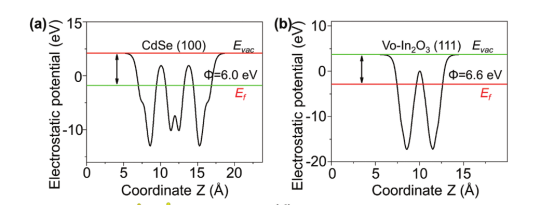
A: 正常的,两侧不对称
Q5:朱老师,计算过渡态(初、末态模型表示为①②),力收敛过程存在负值,然后我看了一下结构,这个负值结构做末态可能更好(将CONTCAR模型表示为③),然后用模型①③做过渡态可以搜出出来。现在问题是我是直接用③替换②,在CONTCAR基础上算scf、freq0;还是需要将③的结构进行二次结构优化,然后再算scf、freq0。后者我试了一下,二次结构优化后的CONTCAR模型与②相似。
A:用3的往下算就可以
Q6:朱老师,体相磁矩为0 按照筛好的自旋组合优化表面模型 出现明显磁矩 算正常吗
A:正常,表面有未配对电子
Q7:朱老师,那表面构型优化时,是不是直接采用体相筛出的自旋组合即可,不用再全部尝试一遍?
A:不用了,用最稳定的
Q8:朱老师,我想问一下就是吸附原子位置放置时需要考虑放置位点金属原子的自旋方向不
A:用体相的就可以,去优化
Q9:朱老师,我想问的是如果ni19和ni9一个自旋向上一个向下,那吸附的时候要考虑把氢原子放在这两个原子上都试一下吗?
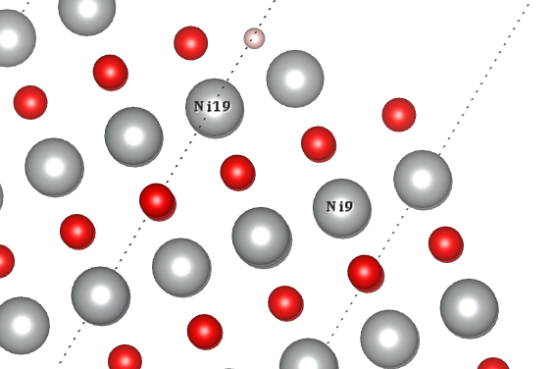
A:一般区别不大,不过可以试试
Q10:老师我做的所有自旋组合的能量差异很小,是不是考虑自旋意义不大
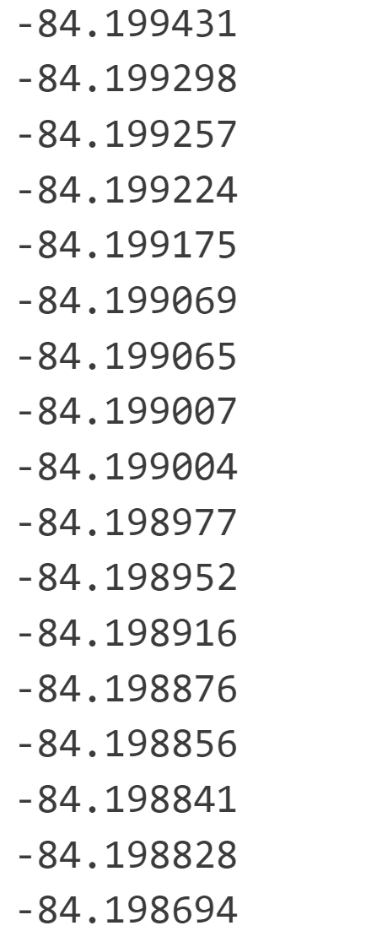
A:吸附时自旋一定要考虑的,磁矩可以用默认
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???