

Q1:朱老师,怎么算氢吸附自由能?
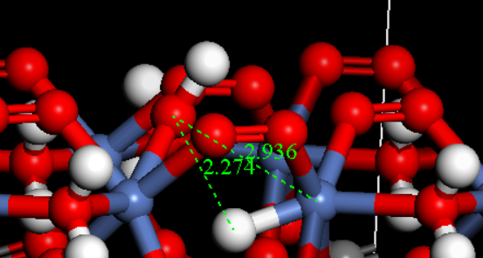

A:没有问题,这样可以直接拿去优化;
标准流程:基态结构→构建 H*→几何优化→能量处理→自由能修正→考虑电位与 pH;
研究“水解离/Volmer 机理”,单独构建 *OH 并保持电荷平衡。
Q2:朱老师,结构优化这个表面模型,接近表面的Na很容易散架?
A:算振动频率,没有虚频;
判断是不是局部极小值的标准只有一个:哈密顿量在该点的二阶导(赫塞矩阵)正定。
在计算里等价为无虚频。
Q3:朱老师,我想计算这个三元正极表面的O原子析出能垒,建立了这样一个表面模型,发现结构优化这个表面模型,接近表面的Na很容易散架,想麻烦朱老师看看,给点调整的建议。
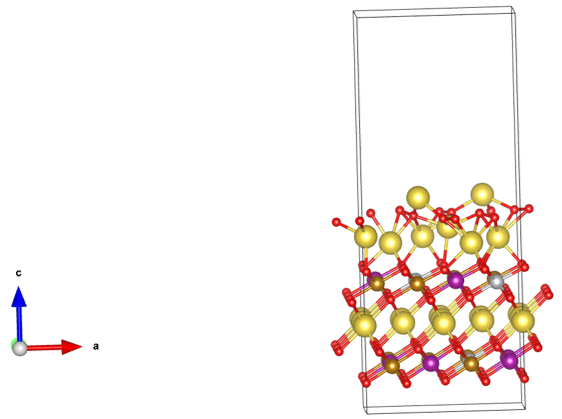
A:这个可以选择让Na不暴露,让TM暴露;
这个 slab 顶层有 Na(或Li)暴露在真空一侧,而且是单面终止。
这会带来
(i)顶面强欠配位、
(ii)净偶极与人造电场、
(iii)局部过氧化/电荷不平衡。
结果就是表面 Na 首先散架/外逸/侧向迁移,优化发散或非常慢。这在层状正极表面是常见坑。
Q4:朱老师,表面模型现在优化失败了?
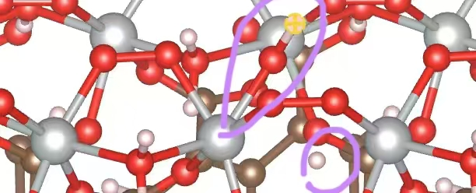
A:离开的远一些;
如果一直会优化回去,那就说这样更稳定,限制不合理;
体系在当前电荷/位点上,拆水后的 H + OH 不是稳定极小值,优化自然往 H2O 分子吸附态滑。
Q5:朱老师,计算甲烷分解过渡态,初态末态都是稳定的,没有虚频,但是计算出来的过渡态能垒很大,十几eV,应该怎么解决呀 ?
A:十几 eV 不是真的很难断 C–H,而是 NEB/TS 路径有问题,可能的原因如下:
原子映射错了,NEB 会把错的原子硬拽到目标位上,途中会出现不可思议的穿插距离,能量飙到十几 eV;
初始带不合理导致中间像重叠,插值把 H 从 C 的背面或穿过金属/氧原子移动;
初末态不是同一反应;
自旋/电子设置不对;
几何/边界条件导致假大能,真空太小、单面 slab 未开偶极修正、原子太近等。
为确保大家的学习成果得以沉淀和复用,我们特将朱老师本次及往期的VASP理论计算答疑精华,系统汇编为【理论计算答疑精选汇编】。它将成为你科研路上随取随用的“速查手册”,助你高效攻克难题。
【做计算 找华算】
华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。