

什么是氧空位?
在理想的金属氧化物晶体中,原子按照严格的化学计量比排列。当晶格中某个氧原子由于热力学涨落、高能粒子辐照或化学还原等原因脱离其晶格格点,便会留下一个空缺,这个空缺即被称为氧空位。
从电荷角度看,一个氧离子(O2-)的离去会留下两个电子,为了维持体系的电中性,这两个电子通常会被束缚在空位附近或转移给邻近的金属阳离子,使其发生价态还原。
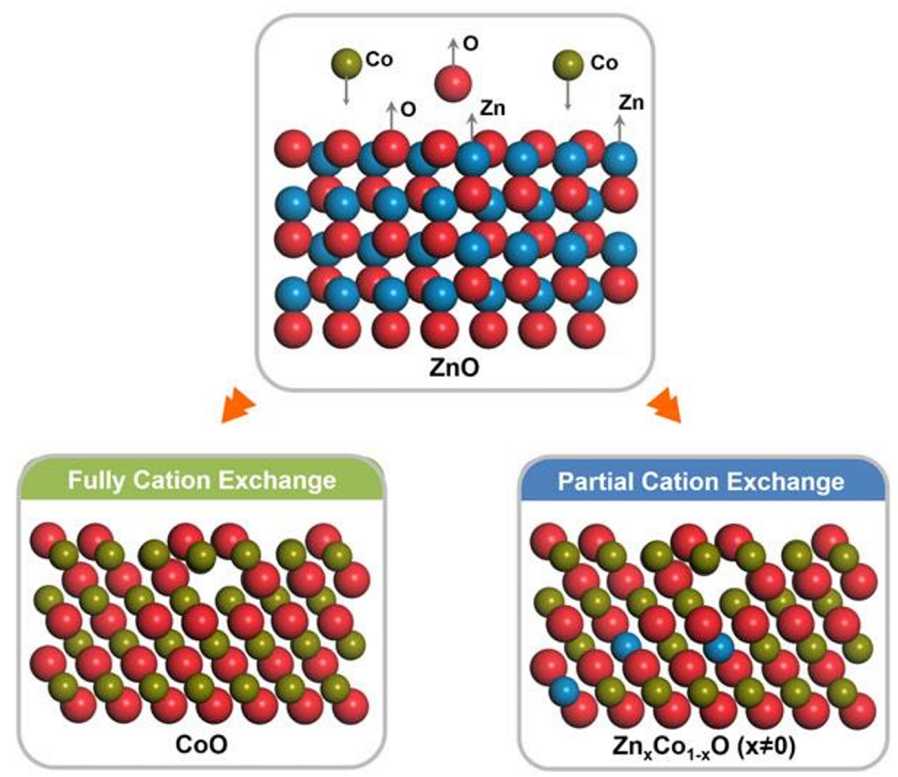

图1 阳离子交换方法合成含有氧空位的ZnxCo1−xO纳米棒。DOI:10.1126/sciadv.aau6261
在Ar、5%H2/Ar和H2气氛中煅烧制备了含氧空位的Sr2Fe1.1Ni0.2Mo0.5O6−δ(SFNM)样品,图2a-c显示了不同SFNM样品的氧空位程度,在H2和5%H2/Ar气氛下,经还原后的SFNM样品的Ni 2p、Fe 2p和Mo 3d光谱的解卷积峰显示出明显的氧化态降低。
这是为了应对表面电荷中性的条件,以获得更热力学稳定的还原结构。图2d显示了不同SFNM样品的O 1s光谱的两个主要解卷积峰,一个位于529 eV的峰对应于晶格氧,第二个位于531 eV的峰对应于吸附氧物种(即O2−、O−),这是氧空位的存在间接识别标志。
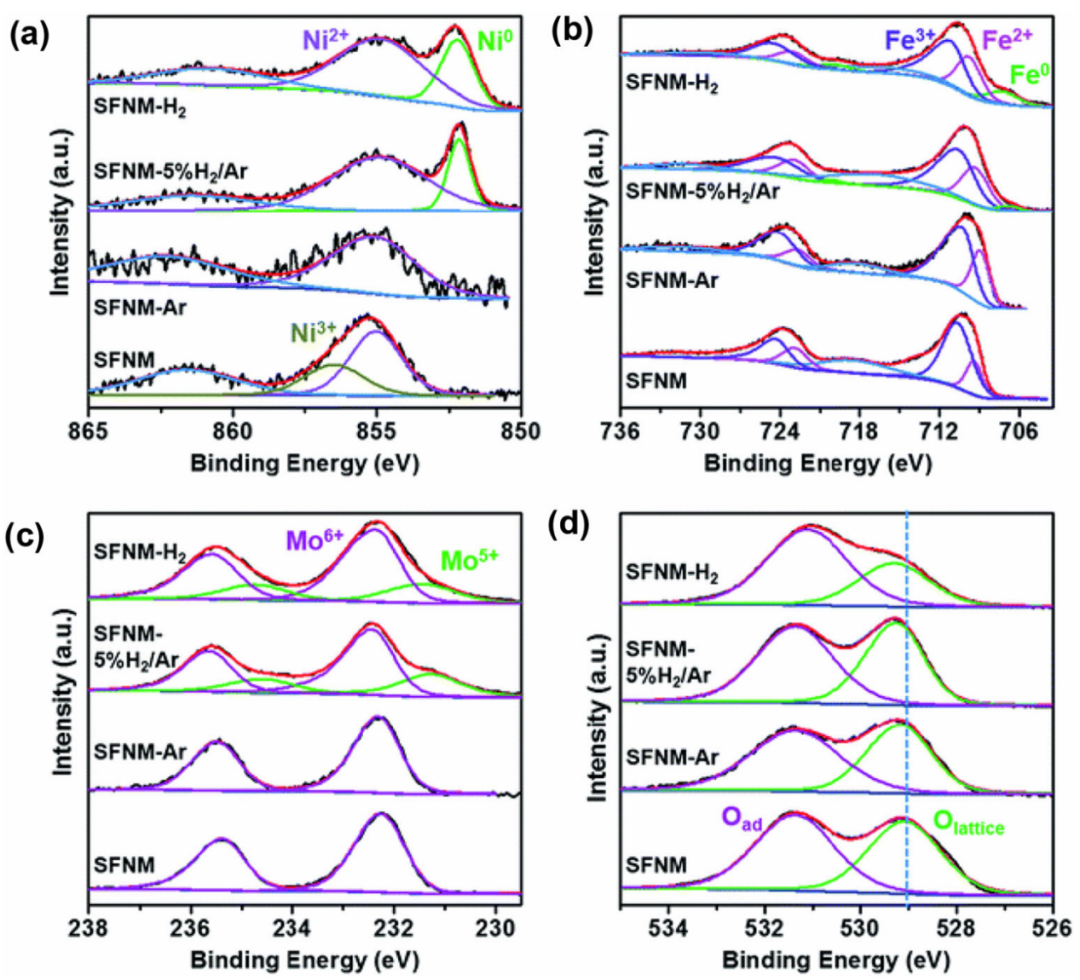
图2 SFNM、SFNM-Ar、SFNM-5% H2/Ar和SFNM-H2的(a)Ni 2p(b)Fe 2p(c)Mo 3d(d)O 1s的XPS光谱。DOI:10.1007/s42247-020-00123-z
XPS分析氧空位的经典误区
一个普遍存在的核心误区是认为XPS可以直接鉴定氧空位并给出一个所谓的“氧空位峰”。然而,XPS的技术原理决定了这是不可能的。XPS测量的是原子内层电子在X射线激发下逃逸出的动能,进而计算出其结合能。空位本身没有原子,更没有电子可供激发和测量。
因此,XPS无法直接探测空位。
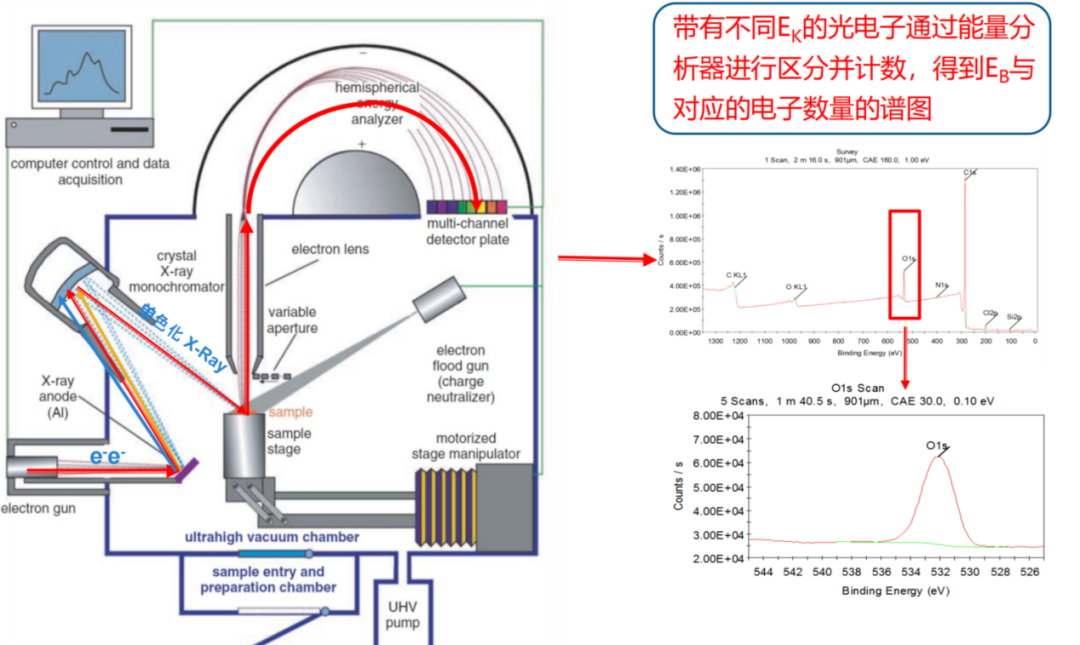
图3 XPS的技术原理示意图
文献中普遍采用的方法是将O 1s光谱反卷积为晶格氧和氧空位组分,后者通常表现出531 eV和532 eV之间的结合能。虽然在文献中被广泛使用,但这一分配和分析没有明确的物理基础。
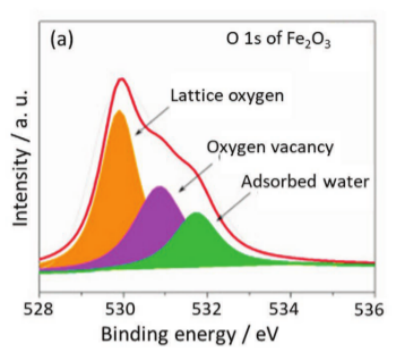
图4 O1s谱分解为晶格氧、氧空位和吸附水。DOI: 10.1002/adfm.202109503
首先,~531 eV特征不能直接由氧空位产生,因为在未被占据的氧位点上不满足XPS信号(氧1s电子的存在)的先决条件。
此外,对CeO2和TiO2的原位XPS研究表明,还原前后O 1s光谱分布基本保持不变,这表明~531 eVO 1s肩峰也不太可能来自氧空位附近的相邻氧位点。相反,越来越多的证据表明,O 1s光谱中的531-532 eV特征来自氧化物表面物理吸附或化学吸附的氧物种(例如羟基)。
由于气体分子可以优先吸附到氧空位上,表面吸附的氧物种和氧空位的浓度有时可能显示出相关性。然而,利用氧吸附物的特征来量化表面氧空位浓度显然是不准确的。例如,即使在完全氧化的样品中,表面吸附的氧浓度也可能很高。因此,依靠O1s光谱中的~531 eV特征来分析氧空位是不准确的。
XPS如何表征氧空位?
尽管如此,XPS依然是表征氧空位的有力工具,其机理在于间接表征。氧空位的存在会对其周围的化学环境产生扰动,这种扰动会通过两种主要途径反映在XPS谱图上。
通过O 1s谱的化学位移进行分析
一个氧空位的形成,会使其周围的氧原子配位数降低,局域电子云密度发生改变,导致这些“氧缺陷区域”(oxygen-deficient regions)的氧原子的O 1s电子结合能相较于完美晶格中的氧原子(晶格氧)发生偏移。
这种能量的偏移,即化学位移,使得原本单一的O 1s峰变得不对称,或可以在分峰拟合后解析出一个新的、位于较高结合能的谱峰。
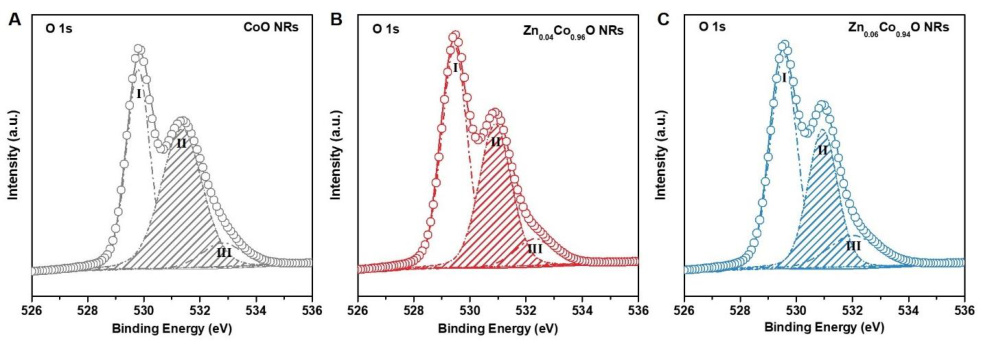
图5 X射线光电子能谱ZnxCo1-xO纳米线的O 1s光谱。DOI:10.1126/sciadv.aau6261
圆圈和线条分别表示实验数据和反卷积数据。在A-C中,低结合能峰(峰I)被归因于晶格氧(OL),而中等结合能峰II与较低氧配位数的大量缺陷位点相关,高结合能峰III则与表面吸附水分子有关。
通过金属阳离子谱的价态变化进行分析
为了维持电中性,氧空位的形成常常伴随着邻近金属阳离子的还原。例如,在TiO2中可能形成Ti³⁺,在ZnO中形成低价态的Znᵟ⁺,在CeO₂中形成Ce³⁺。这些低价态金属离子的核心能级电子结合能与高价态离子存在显著差异。
通过分析金属阳离子的XPS谱图(如Ti 2p,Zn 2p,Ce 3d),可以清晰地分辨并定量这些还原态物种的相对比例,从而间接推断氧空位的浓度。
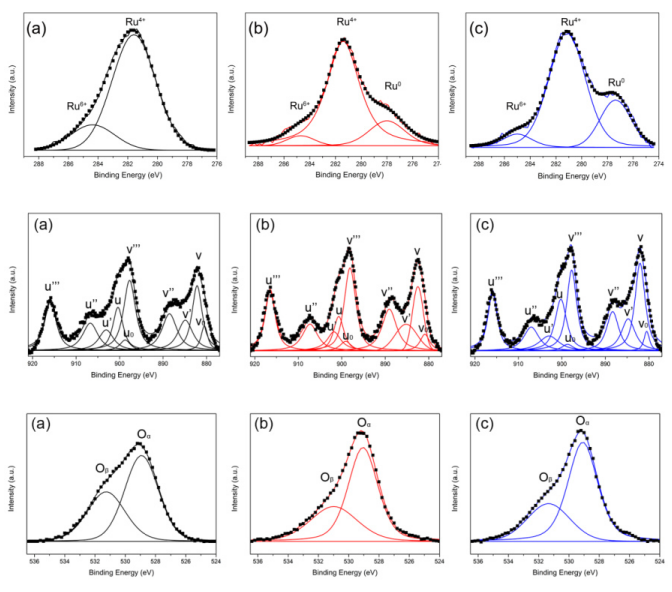
图6 Ru/CeO2-NRs(a)、Ru/CeO2-NCs(b)、Ru/CeO2-NPs(c)的Ru 3d(上)、Ce 3d(中)、O 1s(下)XPS光谱DOI:10.3390/molecules24030526
其中v0、v’、u0、u’这些峰为Ce3+峰,其余为Ce4+峰,Ce3+物种的出现反映了CeO2表面氧空位的形成,高浓度的Ce3+反映了表面氧空位的高度集中。
O 1s的XPS光谱中,在529 eV左右的峰值被归因于CeO2晶格内部的氧物种,标记为Oα物种,而531 eV左右的峰值主要归因于表面低配位氧缺陷。
如表1所示,Ru/CeO2-NRs具有最高的Oβ/Oα比值,而Ru/CeO2-NPs具有最低的Oβ/Oα比值。通过反卷积估计出的Oβ/Oα比值表明,CeO2上的表面氧缺陷含量与形态有关。
表1 Ru/CeO2-NR、Ru/CeO2-NC和Ru/CeO2-NP样品的XPS数据。DOI:10.3390/molecules24030526

XPS如何定量氧空位?
首先,通过分析氧化还原活性阳离子(在氧空位形成时降低其价态),可以确定氧空位的浓度(图7a)。
其次,氧空位可以通过检测表面氧–阳离子化学计量来量化,随着材料释放氧气并形成氧空位,氧空位会减少(图7b)。
第三,由于电子掺杂,在氧空位形成时,氧化物中的电子化学势可能增加。因此,XPS可以通过评估样品内电子化学势的变化来探测材料中的氧空位(图7c)。
需要注意的是,并非三种方法对所有材料都同样有效,因此,为了选择正确的方法,了解材料的物理、化学和光谱行为是很重要的。
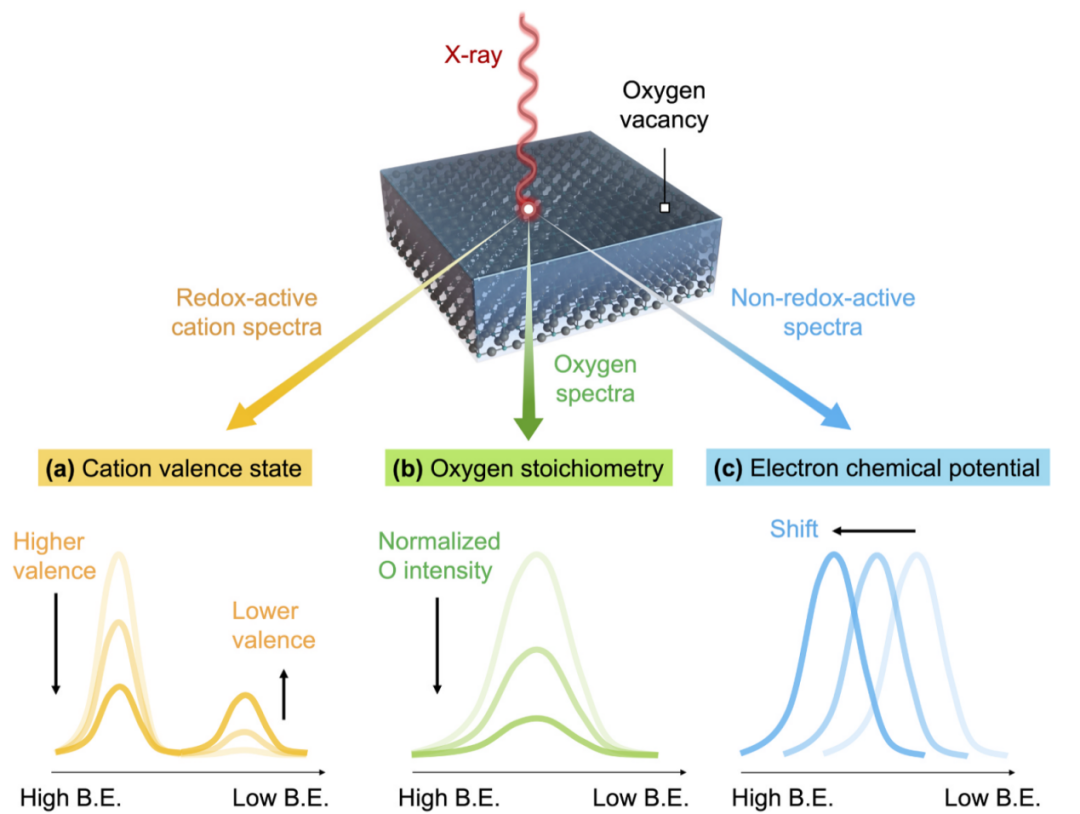
图7 提出了用XPS定量氧空位的三种方法:(a)阳离子价态(b)氧非化学计量(c)电子化学势移DOI:10.1016/j.jeurceramsoc.2024.116709
通过阳离子价态探测氧空位
在氧化物中形成氧空位时,常伴随着氧化还原活性阳离子价态的降低。如果该阳离子的XPS谱图易于解析,便可通过其谱图变化推断氧空位的存在。这一方法尤其适用于阳离子还原态在最外层仅含单电子的情况,此时谱图不易受多重态效应干扰,且用于补偿氧空位的电子局域性较强。
以TiO2为例,氧空位的形成伴随着Ti⁴⁺(3d⁰)还原为Ti³⁺(3d¹),因此在还原态TiO2的Ti 2p XPS谱图中,可在低结合能区观察到Ti³⁺信号。这类体系的配体–金属电荷转移较弱,使阳离子芯能级XPS成为分析价态变化的有效手段。
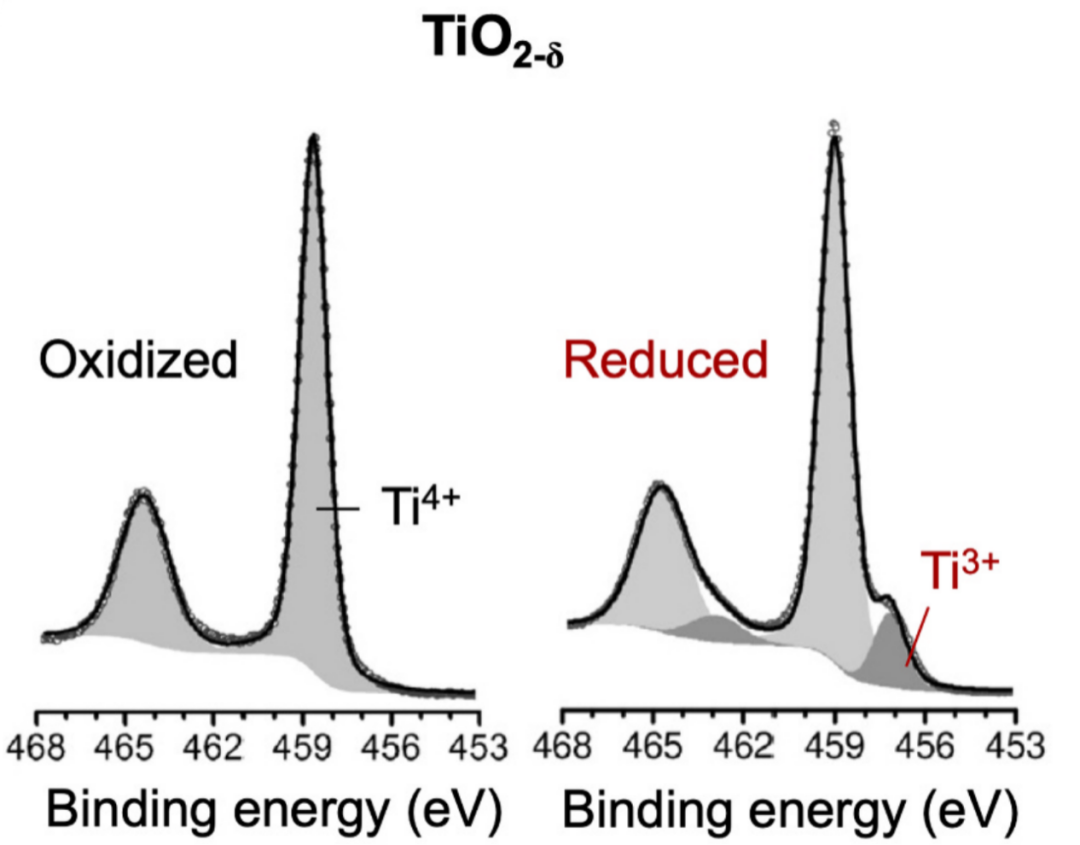
图8 化学计量和还原TiO2的Ti2p光谱DOI:10.1038/s41598-019-48837-3
基于上述规律,可通过XPS定量分析阳离子价态以计算氧空位浓度,其前提是:在XPS探测深度内材料保持电荷中性,且氧空位的电荷补偿完全由电子转移实现,而非通过间隙原子或肖特基缺陷等涉及离子迁移的机制。
例如,在还原态TiO2中,通过拟合Ti 2p谱图可计算出Ti³⁺浓度为12%,进而推得表面组成为Ti⁴⁺0.88Ti³⁺0.12O1.94,对应的氧非化学计量值为0.06。该方法的准确性高度依赖于对XPS谱图的精细拟合。
然而需要指出,并非所有金属氧化物中氧空位的形成都会引起阳离子XPS谱图的显著变化。
例如在负电荷转移氧化物(如La₁₋ₓSrₓFeO₃,LSF)中,氧化还原中心主要位于氧配体上。这类材料在还原过程中,电子组态从3d⁵L(L表示配体空穴)转变为3d⁵,阳离子价态基本不变,因此仅依靠阳离子XPS谱图分析难以有效定量其中的氧空位。
通过氧非化学计量法探测氧空位
当材料释放氧气形成氧空位时,XPS光谱中晶格氧信号会相应减弱,据此可通过量化氧非化学计量(δ)来探测氧空位。具体可采用以下公式计算表面氧非化学计量变化(Δδ):
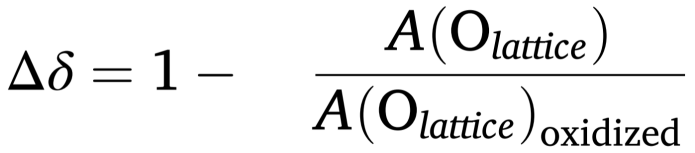
A表示归一化后的晶格氧峰面积(如O 1s),“oxidized”代表氧化态样品。
归一化是必要步骤,因为原始XPS强度易受表面粗糙度、温度、气体氛围等干扰。建议将氧谱强度归一化至氧化还原惰性阳离子谱图的强度(应在同次实验中采集)。
若材料无此类阳离子(如二元氧化物),也可采用XPS本底强度或氧化还原活性阳离子谱图作为参比,但需注意后者可能因反应过程中的波动而引入误差。此外,若氧空位形成伴随表面阳离子偏析或溶出等异质性现象,归一化处理将更为复杂,可能需借助模型辅助分析。
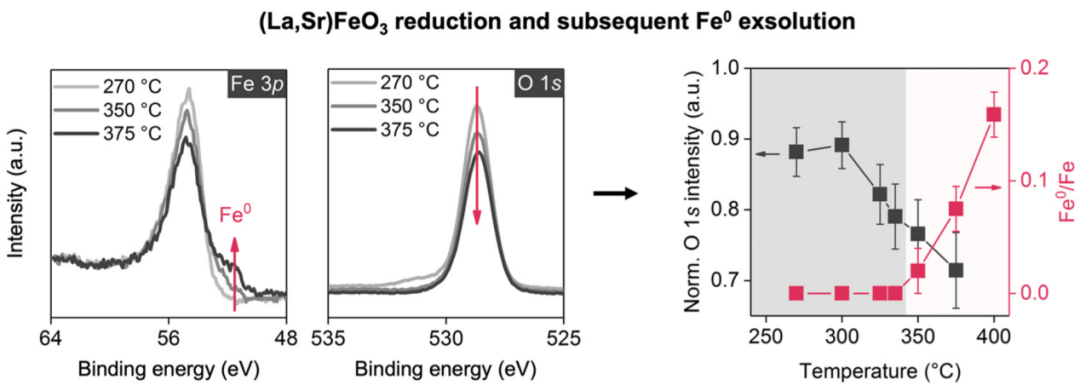
图9 La0.6Sr0.4FeO3-δ在氢气氛围中加热时的原位Fe 3p与O 1s谱图。DOI:10.1021/acs.chemmater.1c00821
图9通过归一化O 1s强度,原位监测了钙钛矿氧化物LSF在H₂中加热时的表面氧化学计量变化。研究采用化学惰性的La 4d谱峰强度作为基准对O 1s进行归一化。
结果显示,归一化后的O 1s强度在300°C以上开始显著下降,表明氧空位在此温度下开始大量形成。
随着温度继续升高,氧强度持续降低,反映了氧空位浓度的不断增加。当温度超过350°C时,谱图中出现金属态Fe⁰信号,这表明材料的氧非化学计量值已超出其钙钛矿相的稳定极限,导致了部分分解。
该案例表明,通过同步分析阴、阳离子XPS谱图,可以有效探究氧空位形成及其诱导的动态相变过程。
此外,通过归一化氧强度变化与表面阳离子浓度变化之比精确量化氧化学计量,也可以准确评估样品中的氧空位浓度。
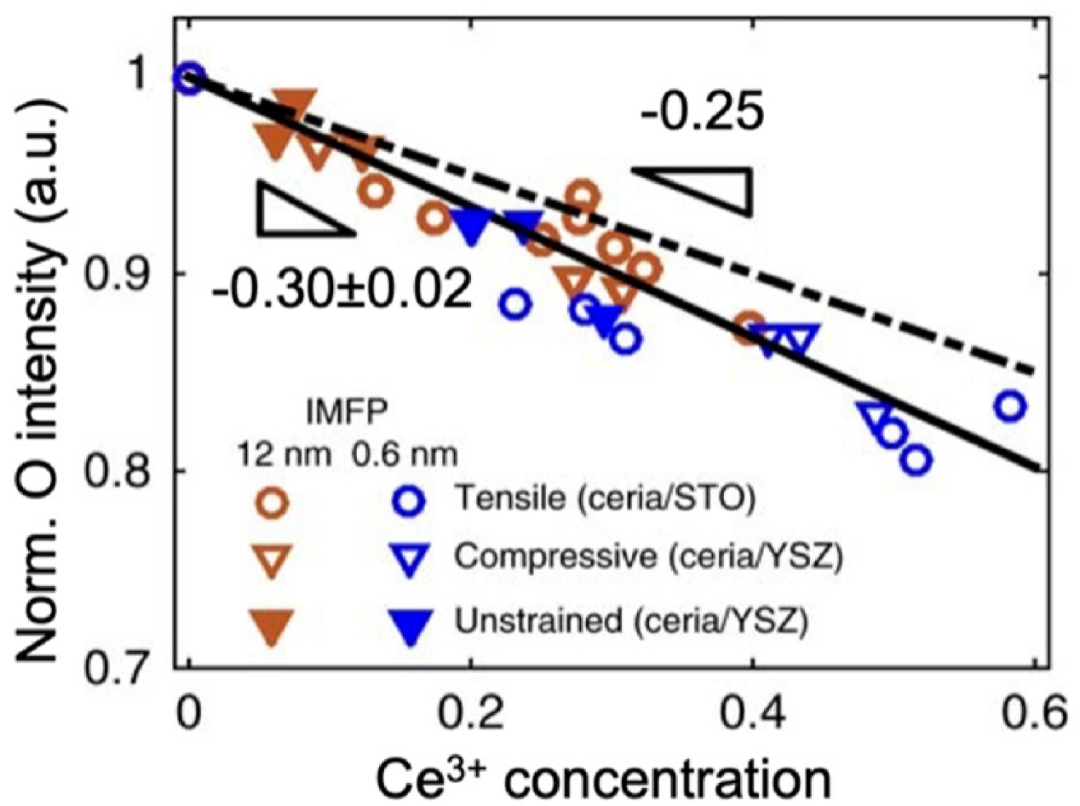
图10 归一化氧强度随着CeO2表面经历还原而下降(实验观测到的氧信号强度与表面Ce³⁺浓度变化之间的斜率为0.30,与理论电中性位点分数0.25高度吻合)DOI:10.1038/ncomms15360
通过费米能级漂移分析探测氧空位
金属氧化物中氧空位的形成常伴随电子掺杂至材料内部,因此费米能级在氧空位形成过程中通常会上移。这一效应同样可通过XPS技术进行研究——因为结合能反映了占据电子态与费米能级之间的能量差,费米能级的上移将导致表观结合能增加。
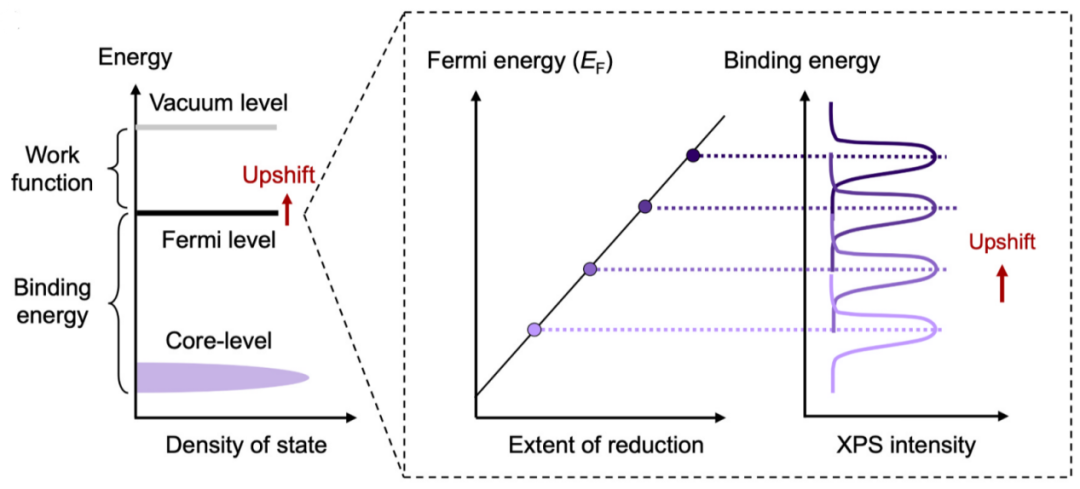
图11 费米能量的变化如何导致XPS结合能移动。DOI:10.1016/j.jeurceramsoc.2024.116709
因此,通过量化XPS结合能的静电位移可以探测氧空位的存在。需要重点说明的是,氧化态变化同样会引起结合能位移(即“化学位移“)。故必须对氧化还原惰性谱线进行深入分析,以确认观测到的结合能位移确实源于氧空位的形成或湮灭。
图12为LSF和STF在615°C氧化与还原气氛下的Sr 3d、O 1s和Fe 2p XPS谱图。Fe 2p谱图显示当暴露于H₂/H₂O气氛时,LSF与STF中铁的价态均明显降低,具体表现为Fe³⁺卫星峰特征减弱。
在整个还原过程中,Sr²⁺和O²⁻保持氧化还原惰性且价态恒定,但Sr 3d和O 1s谱图的结合能在从O2气氛转换至H₂/H₂O还原气氛后出现约1 eV的增长,说明其向高结合能变化的现象并非来自价态变化,而是氧空位形成导致的费米能级上移现象。
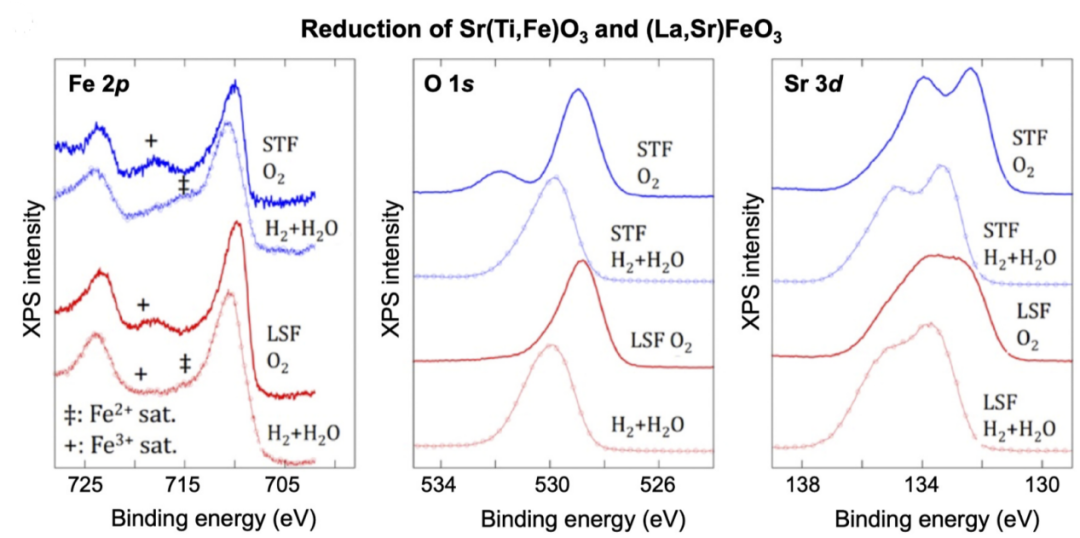
图12 在O2和H2/H2O气氛下STF和LSF之间Fe2p、O1s和Sr 3d光谱的比较DOI:10.1021/acs.jpcc.5b08596
目前通过XPS谱图中静电结合能位移来定量分析氧空位浓度的完整方法体系尚未完全建立,这部分归因于解析复杂结合能位移存在的挑战,但费米能级位移对某些体系中氧空位浓度的微小变化具有响应灵敏度。
XPS如何结合同步辐射?
虽然XPS是表征氧空位的有力工具,但其间接表征的特性以及谱图解析中存在的挑战,使其在精确指认和绝对定量方面仍存在局限。为了更全面、精准地解析氧空位的电子结构与局域配位环境,需要将XPS与同步辐射技术(如X射线吸收精细结构谱XAFS)相结合。
同步辐射技术能够提供元素特异的价态、近邻配位信息,与XPS在表面化学态分析上形成有力互补。
如下图中Nature子刊使用XPS和同步辐射XAS协同表征富氧空位Co3O4材料用于高效CRR(CO2 reduction reaction)。二者联用,可以相互验证,从体相到表面、从静态到动态,共同构建关于氧空位更完整、更可靠的认识,从而将研究推向更深层次。
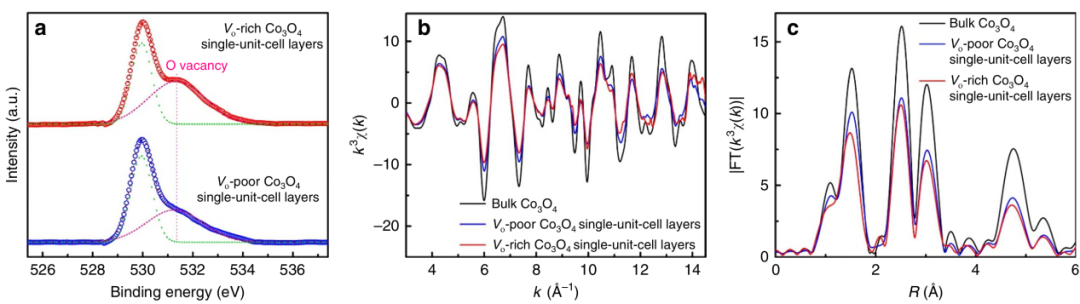
图13 XPS和同步辐射XAS协同表征氧空位材料。DOI:10.1038/ncomms14503
【高端测试 找华算】
华算科技是专业的科研解决方案服务商,精于高端测试。拥有10余年球差电镜拍摄经验与同步辐射三代光源全球机时,500+博士/博士后团队护航,保质保量!
?已助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果在Nature&Science正刊及子刊、Angew、AFM、JACS等顶级期刊发表!
?立即预约,抢占发表先机!