


说明:同步辐射通过原位XAS与SR-FTIR等技术,在原子尺度动态追踪CO₂RR中Cu价态、配位结构及中间体演变,揭示Cu₂-CuN₃团簇为乙醇生成的关键活性位,突破了传统认知,为理性设计高效电催化剂提供依据。
同步辐射是电子在高速曲线运动中产生的一种电磁波,最早于1947年在美国通用电气公司的一台同步加速器中被观测到。
目前,电子储存环被广泛用于为用户提供从红外、可见光、真空紫外、软X射线、中能X射线到硬X射线的连续可调光子能量,覆盖非常宽广的光谱范围。整个波长范围已被用于通过原位/工况实验研究催化材料。
与实验室X射线光源相比,同步辐射光源具有亮度高、通量大、偏振度高以及在极宽范围内连续可调等显著优势。
同步辐射X射线实验方法可分为吸收、衍射、散射、光电子和荧光等几类,在解析电子结构、局域配位环境、晶体结构和表面化学成分等信息方面具有极大优势。
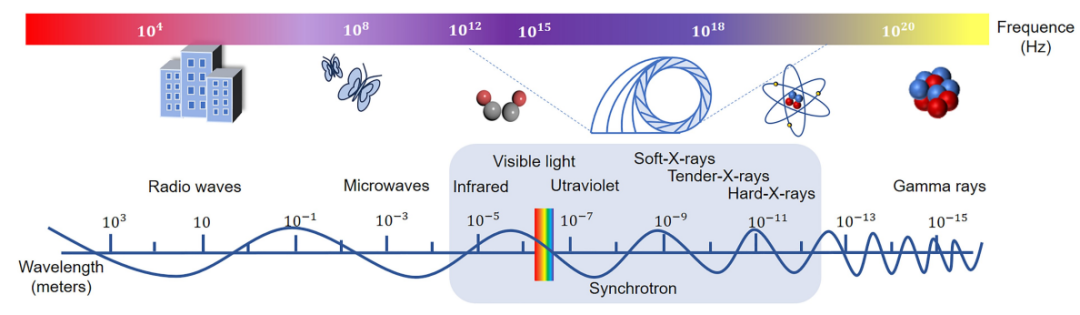
XAS被公认为解析材料中特定元素局域结构和氧化态的强有力工具。当入射X射线能量扫过待测元素内壳层电子的激发阈值时,吸收系数急剧跃升,形成所谓的“吸收边”。
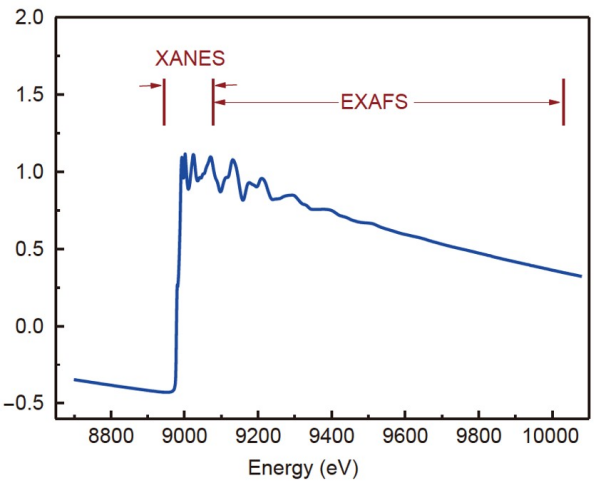
完整的XAS谱图由两部分组成:X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)。XANES对应吸收边上方约30-50 eV范围内的谱区;在此之上的振荡区间则称为EXAFS区域。
在电催化CO₂还原反应(CO₂RR)中,电子结构调控是提升催化性能的核心策略。
其本质是通过调节催化剂表面及活性中心的电子云分布,一方面优化催化剂–电解质界面的电荷传递效率,减少能量损耗;另一方面精准匹配活性中心氧化还原态与CO₂活化(如生成CO₂⁻中间体)、质子耦合电子转移的能级需求。
同时,电子结构改变会直接影响* CO、*HCOO 等中间体的吸附强度——适宜的电子结构能平衡吸附强度,避免过强阻碍脱附或过弱难以稳定中间体,最终构筑低能垒活性位,提升 CO₂RR 的活性、选择性与稳定性。
要动态追踪“电子结构调控–电荷转移-CO₂活化”全过程,同步辐射X射线吸收光谱(XAS)技术凭借原子尺度解析能力成为关键工具,二者功能互补,搭建起 “电子结构–几何结构–催化性能”的关联桥梁:
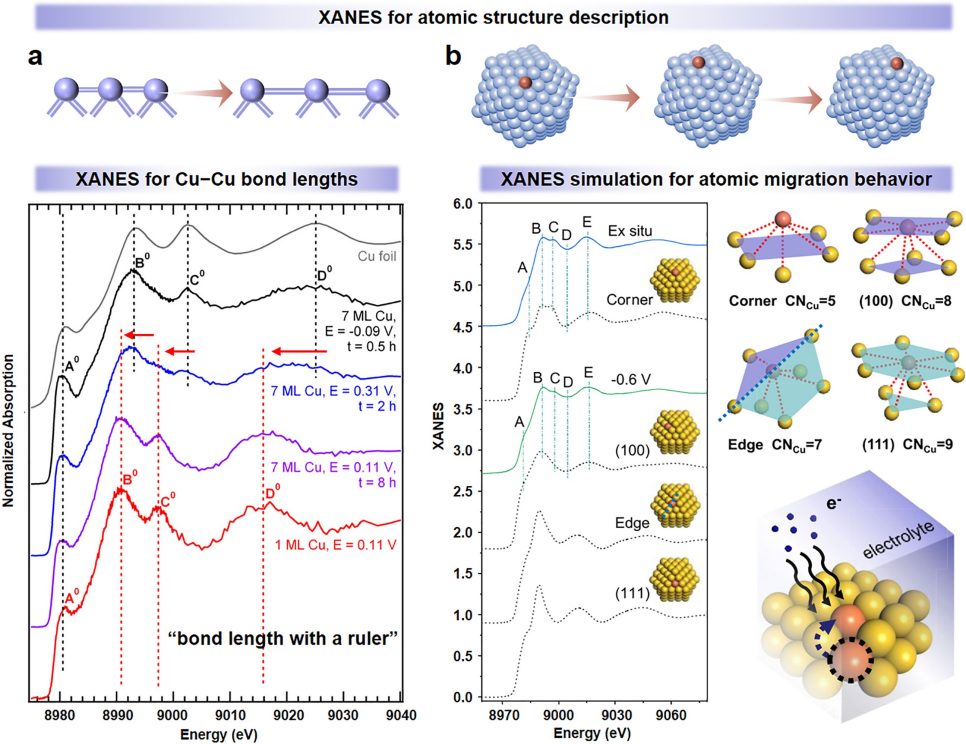
一方面,XANES谱对电子结构变化极具灵敏度。其原理是活性中心原子价电子分布影响内层电子结合能,价态仅变1个电子(如Cu²⁺→Cu⁺),吸收边就会位移数电子伏特。
这种位移直接关联氧化态变化、电荷转移方向(如Cu向N掺杂碳载体转移电子)与转移量,可实时捕捉CO2RR 中活性中心电子结构演变,判断不同电位下活性原子的氧化 / 还原状态及电荷转移是否匹配CO₂活化需求。
另一方面,EXAFS谱是解析局域几何结构的核心手段。
相比XANES,它对近邻原子排布更敏感、空间分辨率达原子级,能穿透宏观形貌与体相结构,提取活性中心的配位数(如Cu周围3个N配位)、近邻原子种类、键长(如Cu-N键 1.93 Å)、晶格畸变及相变信息(如无定形→晶态)。
更关键的是,EXAFS可在外场(如不同电极电位)下实时追踪这些参数,例如监测电位从0 V降至– 1.2 V vs. RHE时,活性中心配位数与键长的变化,反映结构对反应条件的响应。
将 EXAFS实验数据与理论模拟(如FEFF计算、分子动力学)结合,能有效揭示CO₂RR催化剂的“隐藏结构”(如亚纳米团簇、缺陷位点)。
这种“实验+模拟”的方法,可明确反应前后局域几何结构差异,在原子尺度阐明活性位排布如何调控CO₂吸附(线性/弯曲吸附)、活化(C=O键拉长、生成活化中间体)与转化(中间体质子化生成* HCOO/*CO),为理性设计特定局域结构(如特定配位数、键长)的CO₂RR电催化剂提供直接依据,规避传统“试错法”的盲目性。
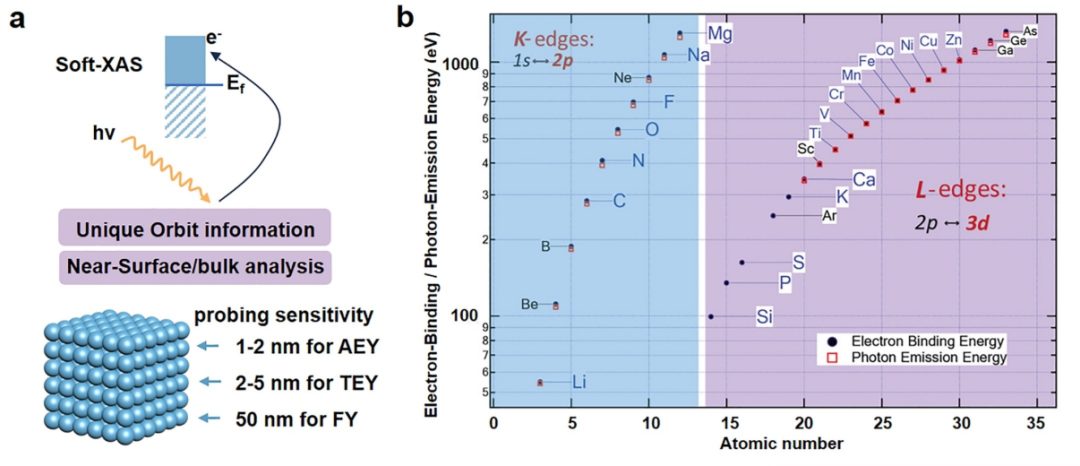
软X射线吸收谱
在XAS技术中,硬X射线(能量通常从几千电子伏到几万电子伏)因其极强的穿透力,非常适合对块体电催化剂进行原位研究。然而,硬XAS主要探测的是不参与催化反应的内层电子。
相比之下,软XAS(能量一般低于5 keV)提供多种表面敏感的探测模式,如俄歇电子产额(AEY,1-2 nm)、总电子产额(TEY,2-5 nm)和荧光产额(FY,约50 nm),各自对应不同的取样深度。
与硬XAS的K边谱相比,软XAS可通过过渡金属的L边直接触及价电子,为CO2电催化中的氧化态变化提供更贴切的视角。
同步辐射的优势在于“高亮度、原位性、多维度”—— 它能在原子尺度上实时追踪催化剂的电子结构、局部几何构型变化,甚至捕捉反应中间体的吸附行为。在本论文(DOI:10.1038/s41467-022-29035-8)中,同步辐射的作用主要体现在三个方面:
原位 XAS:确定Cu2-CuN3电荷不对称团簇
传统认知中,铜基催化剂的活性位点被认为是Cu0纳米颗粒,但这项研究通过同步辐射原位X射线吸收光谱(XAS),推翻了这一假设。
XANES通过吸收边位置判断Cu的氧化态。实验发现,随着外加电位从开路电压(OC)降至-1.1 V vs. RHE,Cu的吸收边逐渐向低能端移动,表明价态从+1~+2逐步降至+0.5左右。
关键转折点出现在– 0.8 V:当电位低于此值时,Cu开始形成金属–金属键(Cu-Cu),但并未完全还原为Cu0(与Cu箔的吸收边位置存在差异)。
EXAFS通过分析散射峰,解析Cu周围的配位环境。
开路状态下,Cu仅存在Cu-O/N配位(键长1.93 Å,配位数3.4),对应分散的CuO团簇;当电位降至-1.1 V(乙醇FE最高时),EXAFS出现新的散射峰(2.4 Å),对应Cu-Cu键(配位数2.0),同时Cu-O/N配位数降至2.3。
结合XANES模拟和冷冻淬灭XAS验证(排除测试过程中的结构变化),团队最终确定:真正的活性位点是“Cu2-CuN3团簇”——2个Cu原子形成金属键,1个 Cu原子与3个石墨型N配位,整体呈现电荷不对称分布。
更重要的是,这种团簇是“电位依赖型”的:一旦撤去电位,Cu-Cu键消失,Cu重新回到Cu-O/N配位状态,证明其是“原位生成的亚稳态活性位点”。
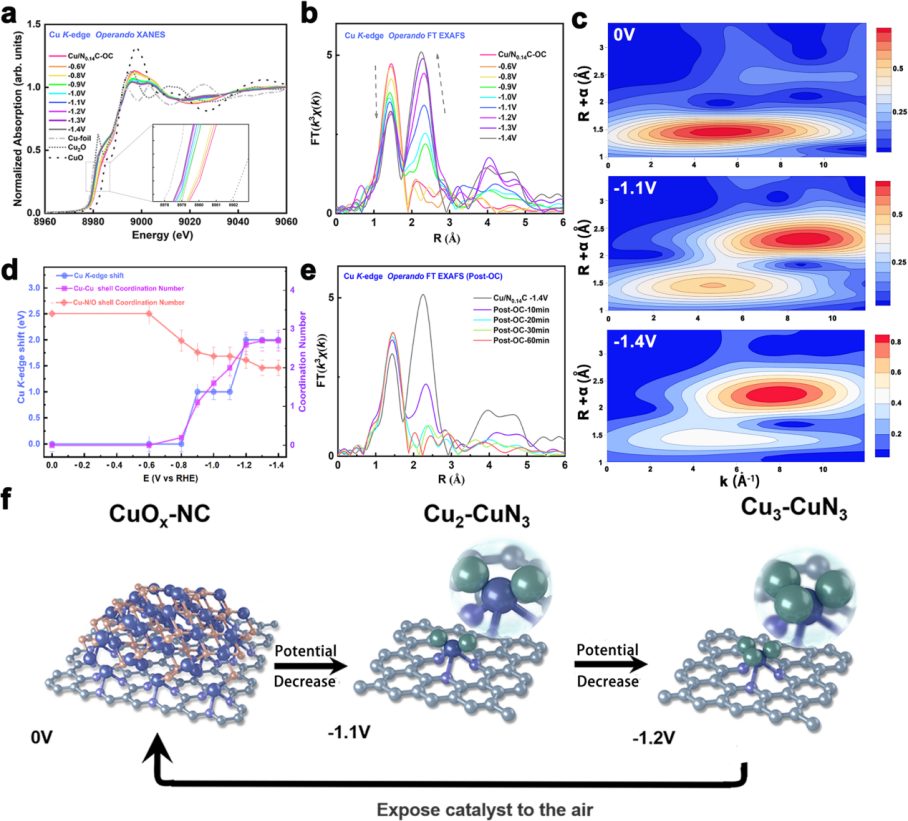
同步辐射SR-FTIR:捕捉反应中间体——CH3*是乙醇生成的关键
CO2RR的选择性取决于中间体的吸附与转化:生成乙醇需要CH₃与OCH₂偶联,而生成乙烯则需要CO*二聚。
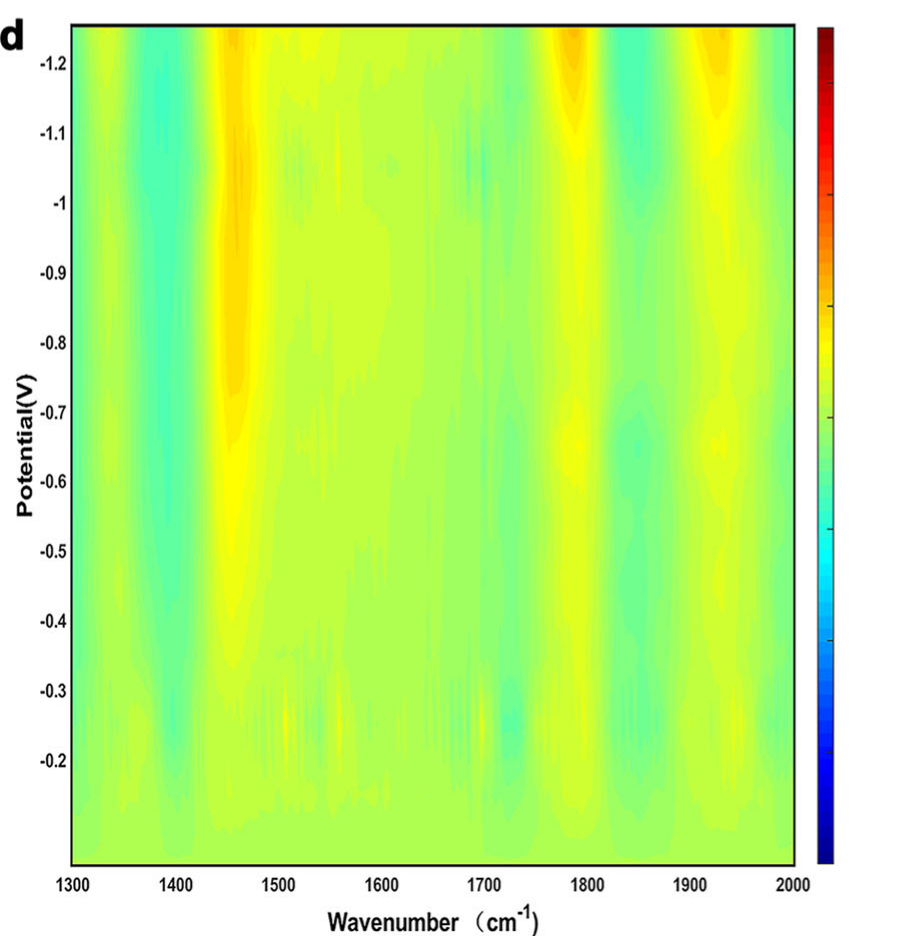
传统红外光谱信号弱、分辨率低,难以捕捉微量中间体,而同步辐射傅里叶变换红外光谱(SR-FTIR) 凭借高亮度优势,成功锁定了关键中间体。
实验发现:当电位低于– 0.7 V时,出现1450 cm⁻¹的特征峰,对应CH3的反对称伸缩振动;当电位降至– 1.1 V(乙醇FE最高)时,该峰强度达到最大;
而当电位进一步降至– 1.2 V,CH3峰减弱,同时出现CO*(2080 cm-1)和碳酸盐(1580 cm-1)的峰,表明此时反应更倾向于生成CO或甲烷,乙醇选择性下降。
这一结果直接证明:CH3*的稳定吸附是乙醇生成的前提,而 Cu2-CuN3团簇的电荷不对称结构,恰好能通过电子富集区域增强CH3的吸附,同时抑制CO二聚,从而导向乙醇生成。
多维度验证:同步辐射与其他表征的协同
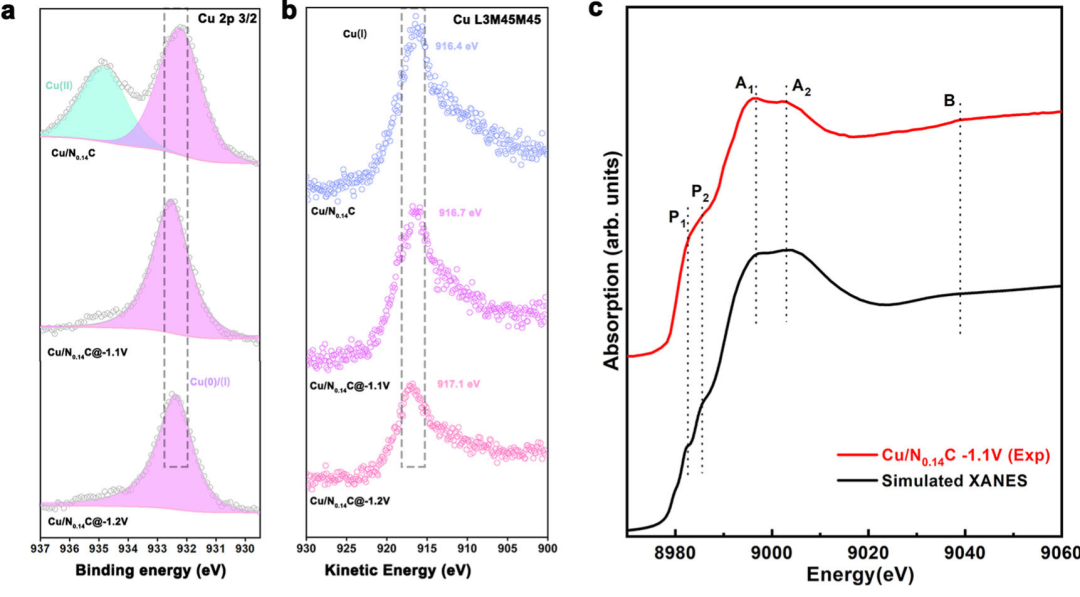
(1)与准原位XPS/AES配合:确认“无Cu0生成”,排除干扰因素。
准原位XPS:通过Cu 2p3/2峰分析价态。当电位降至– 1.1 V时,Cu2+的特征峰(934.6 eV)消失,仅保留Cu+的峰(932.2 eV),未出现Cu0的峰(932.7 eV)。
准原位AES:通过Cu L3M45M45俄歇峰进一步区分Cu+与Cu0。Cu0的俄歇峰位于 918.5 eV,而实验中峰位始终在916.7~917.1 eV,对应Cu+。
两者结果与原位XAS一致,彻底排除了“Cu0纳米颗粒是活性位点”的可能,为Cu2-CuN3团簇的结论提供了独立证据。
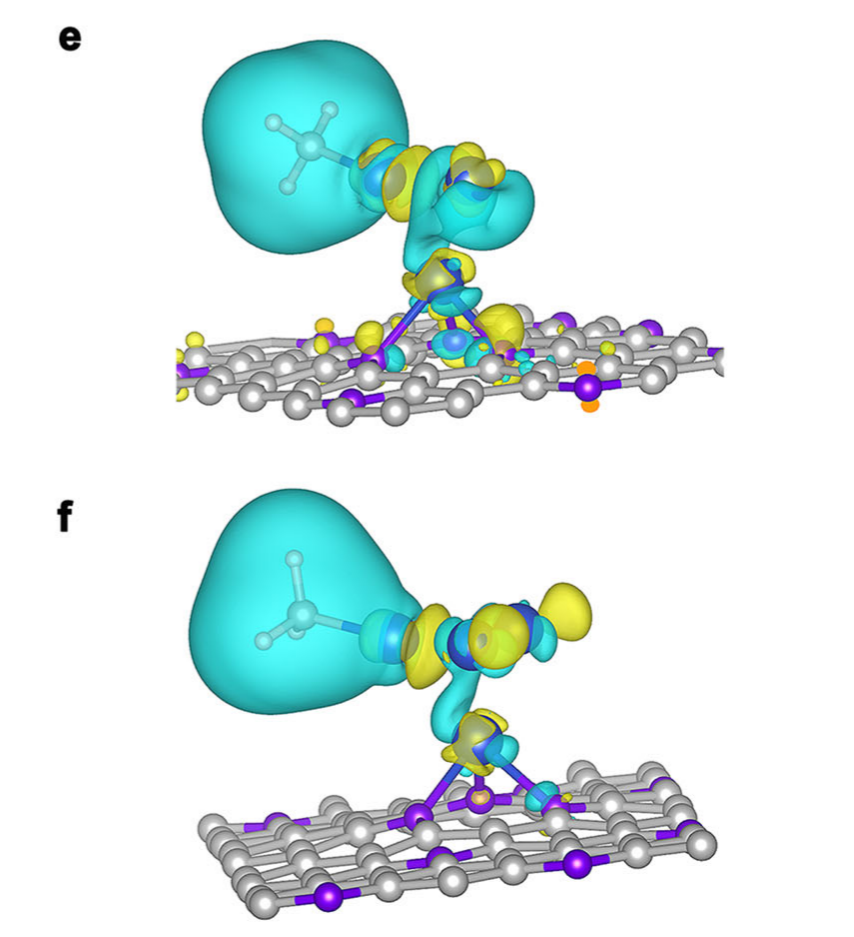
(2)与DFT计算配合:揭示“电荷不对称的作用机制”
计算显示,Cu2-CuN3团簇中,与N配位的Cu原子因N的电负性而失去电子(电荷缺失),另外2个Cu原子则呈现电子富集。这种不对称分布形成局部电场,能增强CO₂的吸附与活化,同时促进CH₃与OCH₂的偶联(能垒降低0.501 eV)。
选择性的“解释器”:对比 Cu₃-CuN₃团簇(电位更负时形成)的计算结果发现,Cu₃团簇对H的吸附能更低(-0.36 eV vs. Cu₂的+ 0.15 eV),导致HER竞争反应增强,乙醇选择性下降。这与同步辐射XAS观察到的 “电位低于– 1.1 V时乙醇 FE下降”完全吻合。
同步辐射技术凭借原位与原子级解析能力,成为CO2制乙醇催化研究的关键工具。
其原位X射线吸收光谱(XAS)通过XANES与EXAFS,精准追踪Cu活性位点动态:从开路时Cu-O/N配位(+1~+2价),到-1.1 V最优电位下形成“Cu2-CuN3电荷不对称团簇”(+0.5价,含Cu-Cu键),直接锁定活性中心,打破 “Cu0为活性位点”的传统认知。
同步辐射傅里叶变换红外光谱(SR-FTIR)则捕捉到CH₃中间体特征信号(1450 cm-1),证实其稳定吸附是乙醇选择性生成的前提,还明确“电位低于– 1.1 V时 CH3减少、CO*增多”的调控规律。
同步辐射与准原位XPS/AES、DFT计算联用,验证Cu价态稳定性(无Cu0)并阐释反应能垒降低机制。虽面临复杂工况模拟、微量位点解析等挑战,但未来随光源分辨率提升、多场耦合装置开发,将助力高性能催化剂设计,支撑碳资源化利用。
【高端测试 找华算】
华算科技是专业的科研解决方案服务商,精于高端测试。拥有10余年球差电镜拍摄经验与同步辐射三代光源全球机时,500+博士/博士后团队护航,保质保量!
?已助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果在Nature&Science正刊及子刊、Angew、AFM、JACS等顶级期刊发表!
?立即预约,抢占发表先机!