

说明:这篇文章华算科技系统介绍了缺陷形成能的概念、计算方法及其在半导体材料设计中的关键作用,掌握如何计算缺陷形成能、理解其受化学势与费米能级影响的机制,并学会运用第一性原理工具(如VASP)预测材料电学性质,为材料设计与改性提供理论支撑。
背景介绍
半导体材料的性质深受缺陷和杂质的影响,其电子结构及缺陷特性是决定电输运性质及电导类型等物理性质的关键因素。深入研究缺陷性质有助于指导材料实现n型或p型掺杂调控,以满足电子器件的设计需求。
在此过程中,第一性原理计算扮演着至关重要的角色,能够精确计算点缺陷的形成能及其缺陷能级。
由于点缺陷存在不同电荷态且其形成能各异,通过计算可以确定不同电荷态缺陷的形成能及其对费米能级的依赖关系,进而预测整个体系内缺陷的总浓度,最终实现对半导体载流子浓度和电导性质的预测。
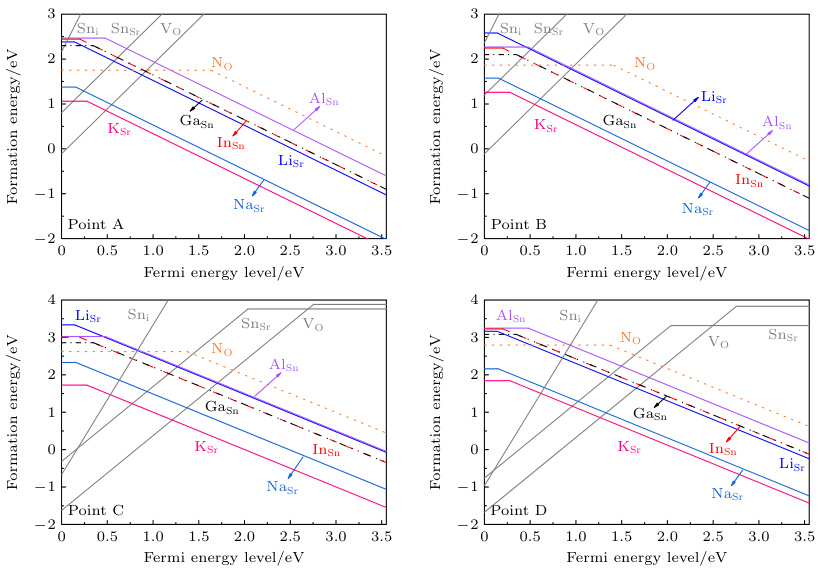

图1 展示不同化学势条件下,Sn、Ga、In、Na等掺杂缺陷的形成能曲线。灰色线条表示可能产生补偿作用的n型本征缺陷形成能。
D0I:10.7498/aps.72.20221544
核心概念
在凝聚态物理和材料科学领域,缺陷形成能是描述在完美晶体中引入一个点缺陷(如空位、间隙原子、反位缺陷、杂质原子等)所需的最小能量。它是一个热力学量,反映了特定缺陷在特定条件下形成的难易程度。
值越低:意味着形成该缺陷在热力学上越有利、越容易自发形成(或在平衡状态下浓度越高)。
值越高:意味着形成该缺陷越困难(或在平衡状态下浓度越低)。
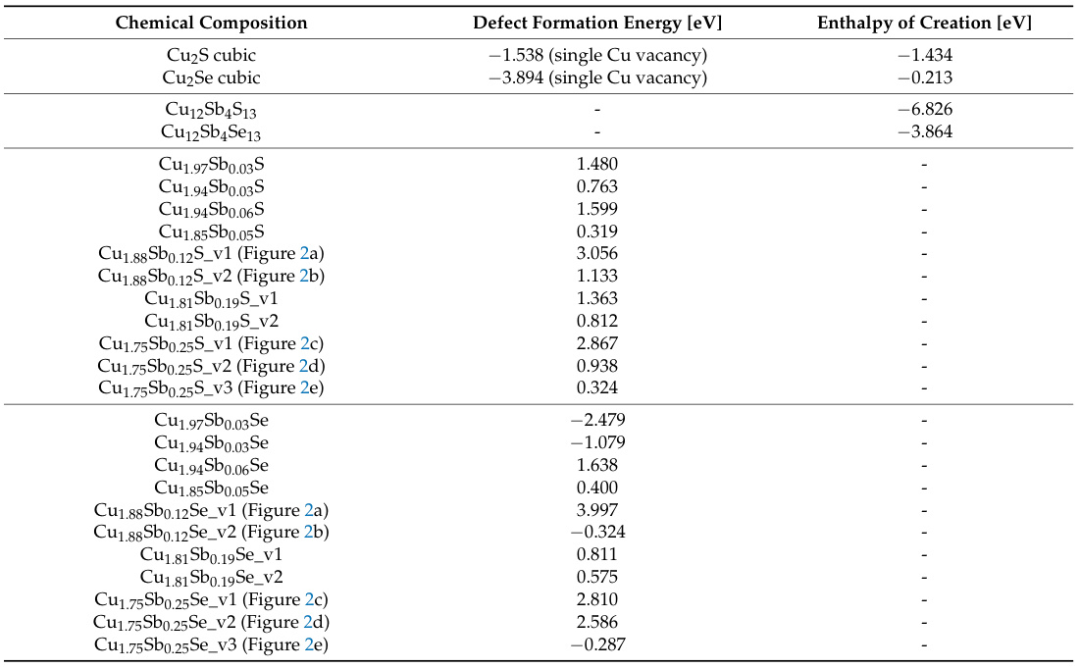
表1 Mikula等(2021)报道Cu₂S和Cu₂Se的立方结构缺陷形成能分别为-1.538 eV和-3.894 eV,负值表明自发缺陷形成
DOI:10.3390/ma14102635
基本计算公式
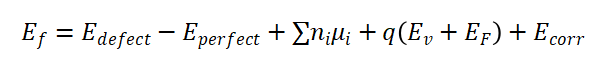
Ef:缺陷形成能;
Edefect:含缺陷的超胞总能量;
Eperfect:完整超胞总能量;
ni:添加(正值)或移除(负值)的原子数;
ui:对应原子的化学势;
q:缺陷电荷态;
Ev:价带顶能量(用于电荷修正);
EF:费米能级
Ecorr:修正项(如静电能修正、有限尺寸修正)
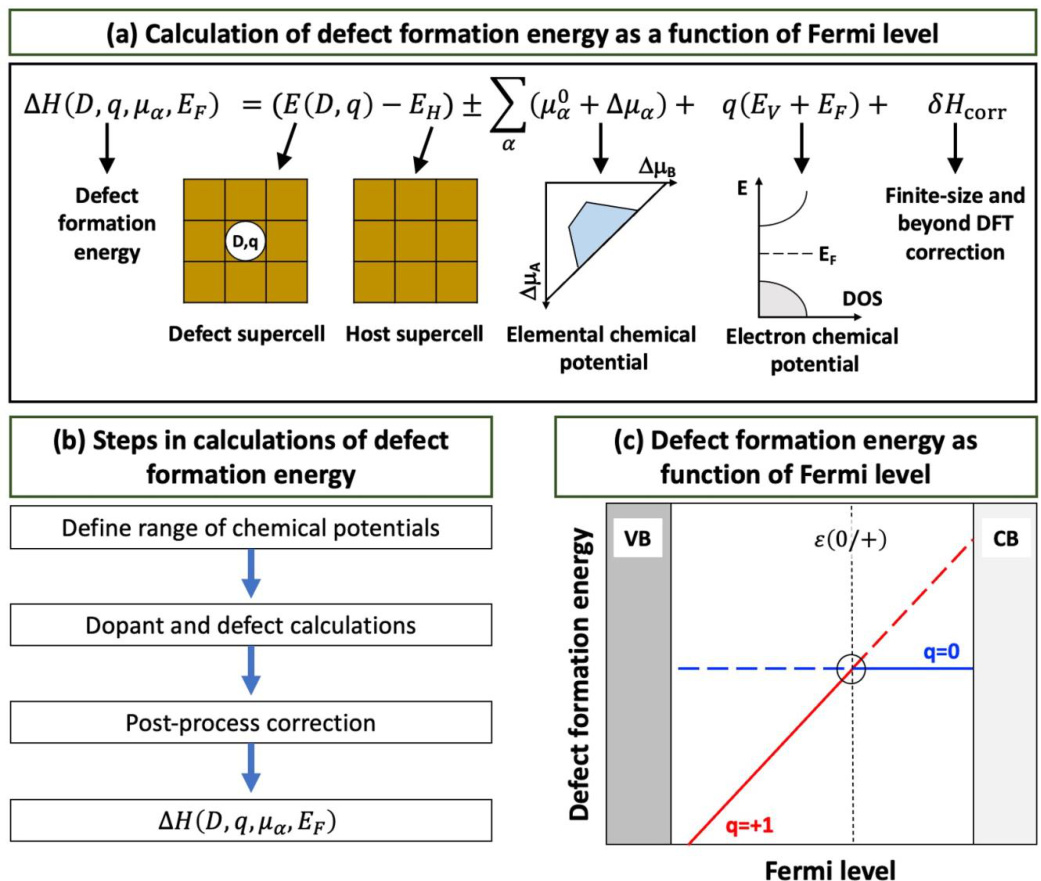
图2 (a)缺陷形成能随费米能级和化学势变化的方程。(b)缺陷形成能计算步骤。(c)缺陷形成能随费米能级和跃迁能级变化的示意图。
DOI:10.1021/acs.chemrev.0c00608
关键影响因素
原子化学势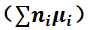
物理意义:这部分代表了在形成缺陷时,系统与外部原子库(或粒子库)交换原子所需的能量成本。ui是原子种类
的化学势,可以理解为该原子在特定环境(温度、压力、组分)下的“能量单价”。ni是形成该缺陷时需要添加(ni>0)或移除(ni<0)的原子i的数量。
关键点:ui的值完全取决于材料所处的热力学环境。
例如富氧环境:向系统中添加氧原子相对容易。因此,形成氧空位的成本很高,形成金属空位或氧间隙的成本较低(需要添加一个氧原子或移除一个金属原子,但移除金属原子成本受uM限制)。
缺氧/富金属环境:向系统中添加氧原子困难。因此,形成氧空位的成本很低,形成金属间隙的成本也可能较低,而形成氧间或金属空位的成本很高。
费米能级![]()
![]()
物理意义:这部分代表了改变缺陷电荷状态所需的电子能量成本。
是缺陷的电荷态(单位:基本电荷e)。EF是费米能级,表示电子化学势(向系统添加或移除一个电子的能量参考点),通常相对于价带顶EVBM给出。
关键点:电荷态的敏感性
对于施主缺陷:当EF升高(系统更容易失去电子),形成能Ef降低(更容易形成带正电的施主缺陷)。
对于受主缺陷:当EF升高,形成能Ef升高(更难形成带负电的受主缺陷)。
对于中性缺陷:形成能不依赖于EF。
缺陷类型和构型
物理意义:不同种类的缺陷(空位、间隙、反位、杂质)或者同种缺陷在不同晶格位置或不同构型(弛豫后的原子结构),其引起的晶格畸变程度、键合断裂/重构情况、电子结构变化都不同,这直接反映在缺陷超胞的总能量Edefect上。
关键点:内在差异
空位:移除一个原子,留下空位,周围原子弛豫。形成能通常与键能、原子尺寸有关。
间隙:在晶格间隙位置插入一个原子,导致局部晶格膨胀和畸变。形成能通常较高,且强烈依赖于间隙位点的类型和大小。
反位:两种不同原子交换位置(在化合物半导体中常见,如GaAs中的As-Ga, Ga-As)。形成能与原子间的化学亲和力、尺寸失配有关。
杂质:外来原子替换晶格原子。形成能取决于杂质原子与被替换原子的相似性(尺寸、电负性、价态)、杂质来源的化学势以及杂质引起的晶格畸变和电荷补偿需求。
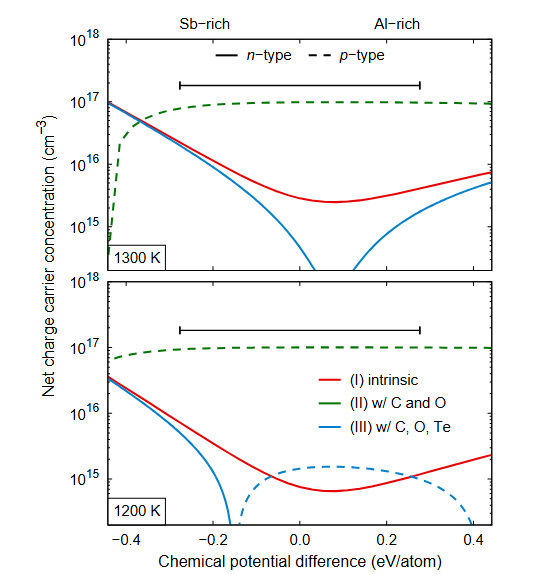
图3 原子缺失形成的空位,如铝空位(V)和锑反位(Sb)在AlSb中形成复合缺陷
DOI:10.1103/PhysRevB.81.195216
计算修正项
物理意义:由于理论计算本身的近似和模型限制(主要是使用周期性边界条件的有限超胞),计算结果需要修正才能更准确地反映真实(无限大)晶体中的情况。
忽略这些修正会导致显著误差,尤其对带电缺陷。前面提到过缺陷分受主类型和施主类型,缺陷能级会从价带得到电子或向导带提供电子,从而成为带电缺陷。
图4 周期性条件下的带电点缺陷计算模型,(a)施主缺陷模型(失去一电子带正电)(b)带正电的周期性超胞间的库伦相互作用(c)施主能级失去的电子跃迁到等效的费米面(d)在超胞中添加的带负电的Jellium背景电荷。
https://www.bilibili.com/video/BV1dU4y1y73k/?p=9&vd_source=fb017bb975ea6
如图4(a)所示,第一性原理计算通过增加或减少体系电子数目来模拟体系带电。在带电体系的计算时,我们已经了解能带偏移是其中一部分误差来源,其实除此还存在更大的误差来源。
如图4(b)所示,周期性边界条件下,带电缺陷与其镜像电荷之间存在静电相互作用,该作用能会随超胞尺寸增大而发散。这也造成我们任意选取的计算超胞带电时的能量并不是真正体系的总能。
有限尺寸修正:
问题:计算带电缺陷时,超胞中的净电荷q会在周期性镜像之间产生强烈的、非物理的(长程)库伦吸引/排斥,显著影响总能量Edefect。
修正方法:
马克-海因 (Makov-Payne) 修正:最经典的方法,将点电荷在周期性边界条件下的静电能量展开,主要修正项与 1/L (L是超胞尺寸) 成正比。
均匀背景电荷+ 其他方法:在计算时添加一个均匀的背景电荷以中和超胞总电荷,然后在后处理中修正背景电荷引入的误差。
带隙修正:
问题:标准局域密度近似(LDA) 或广义梯度近似 (GGA) 的密度泛函理论系统性低估半导体/绝缘体的带隙。这导致缺陷能级在带隙中的位置计算不准,进而影响Ef对EF的依赖关系(尤其是当缺陷能级靠近带边时)。
修正方法:
杂化泛函(Hybrid Functionals, e.g., HSE06):包含部分精确交换,能显著改善带隙预测,但计算量巨大。
GW方法:更高级的准粒子方法,精度高但计算量极其昂贵。
VASP空位形成能计算流程
构建完美晶格模型
创建无缺陷的超晶胞(Supercell),进行充分的结构优化(几何弛豫),确保体系处于能量最低的基态。计算完美晶格的总能量Eperfect(单位:eV),记录原子数N。
构建缺陷模型
移除目标原子形成空位,生成含空位的超晶胞(原子数变为N-1),对含空位的超胞进行几何优化:允许空位周围原子弛豫,以模拟局部结构变化;使用高精度参数(如ENMAX增大20%,KPOINTS网格加密)确保能量收敛;记录优化后的总能量Edefect。
计算空位形成能
基本公式:
此公式直接比较弛豫后缺陷体系与完美晶格的相对能量
带电缺陷修正:
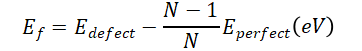
总结
重要性:缺陷形成能是连接材料原子尺度结构(缺陷)与其宏观可观测性质之间的关键桥梁。无论是理解材料的本征行为还是进行有目的性的材料改性,缺陷形成能的计算和分析都是不可或缺的工具。
核心应用:通过量化特定环境下(化学势ui和费米能级EF)创造点缺陷所需的能量,精准预测该缺陷的热力学稳定性、平衡浓度以及主导电荷态,从而理解并调控材料的关键性质(如电导率、光学行为、离子迁移率),为设计高性能功能材料(如半导体、离子导体、催化剂)提供理论依据和优化方向。