

形成能作为热力学稳定性的核心指标,在电催化材料设计中发挥着关键作用。通过密度泛函理论(DFT)计算,研究者可量化化合物与单质间的能量差异,结合凸包图分析筛选热力学稳定相,规避材料失稳风险。
在单原子催化剂设计中,形成能差异揭示了活性位点的稳定性与构效关系,例如Fe-N₄位点因其低形成能成为高效氧还原活性中心。实例分析表明,Pt-O-P结构通过强相互作用抑制纳米颗粒迁移,显著提升催化剂耐久性。
当前研究通过集成机器学习与高通量计算,加速多元材料体系的稳定相筛选,同时结合动态电化学界面修正模型,推动材料设计从经验试错转向数据驱动。未来需进一步解决多尺度耦合挑战,如高温熵效应与界面动态重构的影响,以实现更精准的稳定性预测与催化性能优化。


形成能的定义与物理意义
形成能(Formation Energy)是描述化合物从其组成元素的稳定单质状态形成时能量变化的物理量。在密度泛函理论(DFT)计算中,形成能的计算公式为:
Eform=Ecompound−∑niμi
其中, Ecompound 为化合物的总能量,ni 为化合物中元素 i 的原子数,μi 为元素 i 在稳定单质状态下的化学势。例如,对于二元化合物 AxBy,其形成能可表示为:
Eform=E(AxBy)−xE(Abulk)−yE(Bbulk)
形成能为负值时,表明化合物相对于单质更稳定(放热过程);正值则表示需要外界能量输入才能形成(吸热过程)。
形成能与内聚能的区别:
1. 内聚能(Cohesive Energy)基于自由原子能量计算,反映原子从孤立状态结合成晶体的能量变化。
2. 形成能基于稳定单质的能量,反映化合物与单质之间的能量差,更适用于评估材料的热力学稳定性。
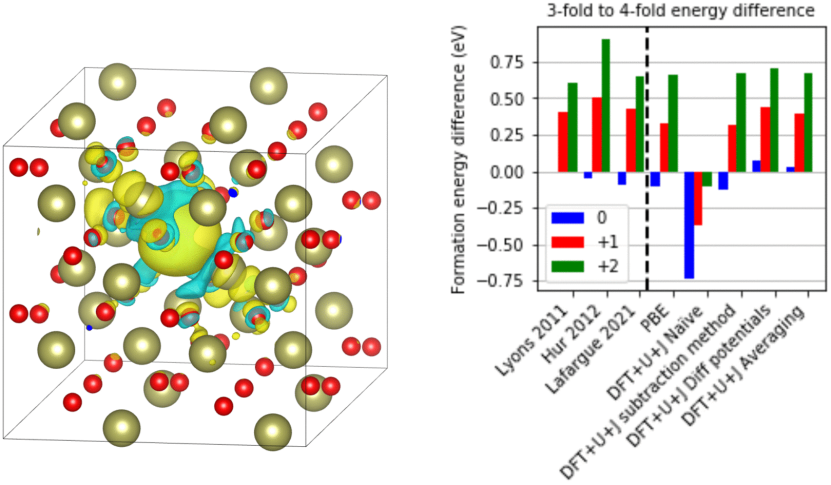
DOI:10.1039/d4ra07774a
电催化计算中形成能的分析方法
在电催化领域,形成能分析主要用于筛选稳定催化剂、预测反应路径及活性位点。
在材料热力学稳定性评估中,凸包图(Convex Hull)作为一种经典的分析工具,通过量化化合物形成能与组分比例的关系,为材料稳定性预测提供了直观判据。
其核心原理基于吉布斯自由能最小化准则:对于特定化学组分体系(如二元合金系统),首先通过密度泛函理论(DFT)计算不同原子比例下各可能相的形成能,随后将计算结果绘制于能量-组分坐标系中,并通过几何方法构建连接最低能量点的凸包边界线(即凸包线)。
若某成分点的形成能位于凸包线以下,表明该组分对应的材料在热力学上处于绝对稳定态;反之,若其位于凸包线上方,则该材料可能以亚稳态存在或具有热力学不稳定性,在特定外界条件(如温度、压力、电化学势场)下可能发生相变或分解。
例如,在开发高活性二元合金催化剂(如Pt-Cu体系)时,研究者通过DFT计算不同Pt/Cu原子比例下合金的形成能,构建凸包图后可筛选出在目标电催化反应电位区间内处于稳定区域的合金相(如Pt₃Cu、PtCu₃等),从而规避实验合成中因热力学失稳导致的元素偏析或结构坍塌风险。
这一方法不仅大幅提升了催化剂的筛选效率,还可通过分析凸包斜率揭示相分离趋势,为设计具有特定表面原子排布的合金催化剂提供理论指导。
值得注意的是,凸包分析需结合动态稳定性评估进行补充,例如通过声子谱计算验证亚稳态材料的动力学稳定性,或引入电化学电位修正项构建电化学凸包图(Electrochemical Convex Hull),以模拟实际工况下电极材料与电解液界面处的化学势变化。
近年来,随着高通量计算与机器学习技术的融合,凸包分析已拓展至三元甚至多元材料体系,通过自动生成成分-能量数据集并结合聚类算法识别稳定相,显著加速了新型催化材料的发现进程。
然而,该方法仍面临挑战,如高温熵效应对凸包结构的修正、表面重构对体相稳定性的影响等,需进一步发展多尺度耦合模型以实现更精准的稳定性预测。
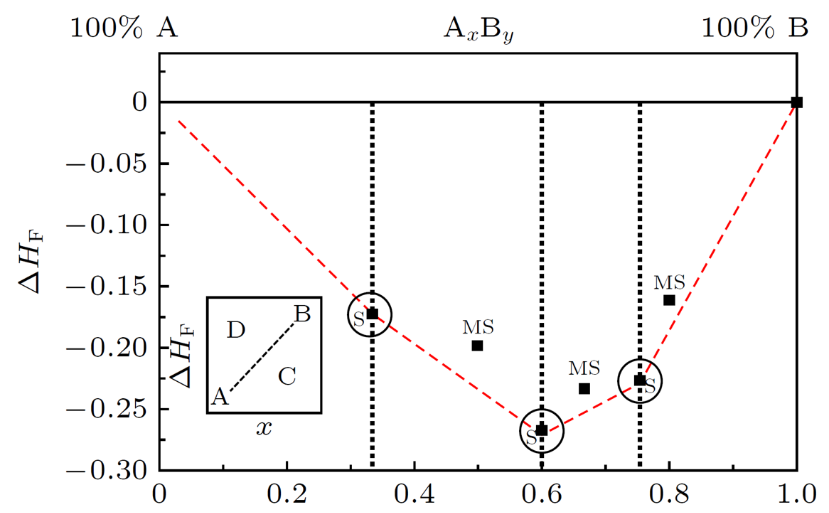
DOI:10.7498/aps.70.20201636
在单原子催化剂的设计与优化中,活性位点形成能计算是揭示其热力学稳定性和催化性能的关键理论工具。
该方法基于密度泛函理论(DFT)模拟,通过量化不同原子配位构型(如Fe-N₄、Fe-N₃O、Fe-S₁N₃等)的形成能差异,筛选出在合成或反应条件下最可能稳定存在的活性中心。
具体而言,形成能定义为将孤立金属原子与载体结合形成特定配位结构所需的能量变化,其计算需考虑金属原子的嵌入能、载体缺陷形成能及配位原子(如N、O、S)的化学势平衡。
例如,在Fe基单原子催化剂中,若Fe-N₄位点的形成能显著低于Fe-N₃O或Fe-C₂N₂等构型,表明Fe原子倾向于与四个氮原子配位形成平面四边形结构,该位点不仅具有热力学稳定性,且在氧还原反应(ORR)中因适中的*OOH吸附能而表现出优异的催化活性。
这一方法已被广泛应用于揭示M-Nₓ(M=Fe, Co, Mn等)位点的构效关系:通过系统计算不同配位数、杂原子掺杂(如O、S、P取代)及载体晶格应变对形成能的影响,可预测活性位点在高温煅烧或电化学极化过程中的结构演化规律,从而指导实验合成中前驱体配体设计与热处理条件的优化。
值得注意的是,形成能分析需结合动力学稳定性评估(如过渡态搜索、分子动力学模拟)以全面评估活性位点的实际存在概率,例如某些高形成能的亚稳态构型可能因动力学势垒较高而在反应中短暂存在并参与催化循环。
此外,环境因素(如pH值、电位、溶剂化效应)可通过修正化学势项被纳入形成能计算框架,例如在酸性电解液中,Fe-N₄位点可能因质子化作用转变为Fe-N₃H⁺构型,此时需重新计算质子化前后的形成能差异以判断其稳定性演变。
近年来,该领域正朝着多尺度耦合方向发展:一方面通过机器学习势函数加速高精度形成能数据库的构建,实现从二元到多元配位结构的快速筛选;另一方面结合原位表征技术(如X射线吸收谱、扫描隧道显微镜)验证计算预测的准确性,形成“计算-实验闭环”的设计范式。
然而,现有方法仍面临挑战,例如载体边缘效应、动态界面重构对局部配位环境的扰动尚未被充分量化,需进一步发展基于动态电化学界面的形成能修正模型,为设计兼具高活性与长寿命的单原子催化剂提供更精准的理论支撑。
DOI:10.1016/j.physb.2025.417017
DFT形成能判断结构稳定性分析

该研究通过密度泛函理论(
DFT)计算探讨了P-O功能基团对PtCo合金纳米颗粒的锚定机制,重点分析了Pt与P-O基团间相互作用的稳定性。
计算构建了两种界面模型:Pt原子通过P-O基团中的P原子直接连接(Pt–P–O)和通过O原子间接连接(Pt–O–P)。
结果表明,Pt–O–P结构的形成能为-2.59 eV,显著低于Pt–P–O结构的-0.73 eV,表明Pt–O–P在热力学上更稳定且优先形成。
这一能量差异源于O原子较高的电负性,使其与Pt的相互作用更强,从而有效抑制纳米颗粒的迁移和奥斯特瓦尔德熟化过程。
此外,退火后的元素分布模拟进一步验证了P-O基团与Pt之间的强亲和力,导致P和O元素富集于Pt表面而非碳载体,强化了锚定效应。
DFT计算从原子尺度揭示了P-O功能基团通过稳定Pt–O–P配位结构提升催化剂耐久性的内在机制,为设计高稳定性电催化剂提供了理论依据。
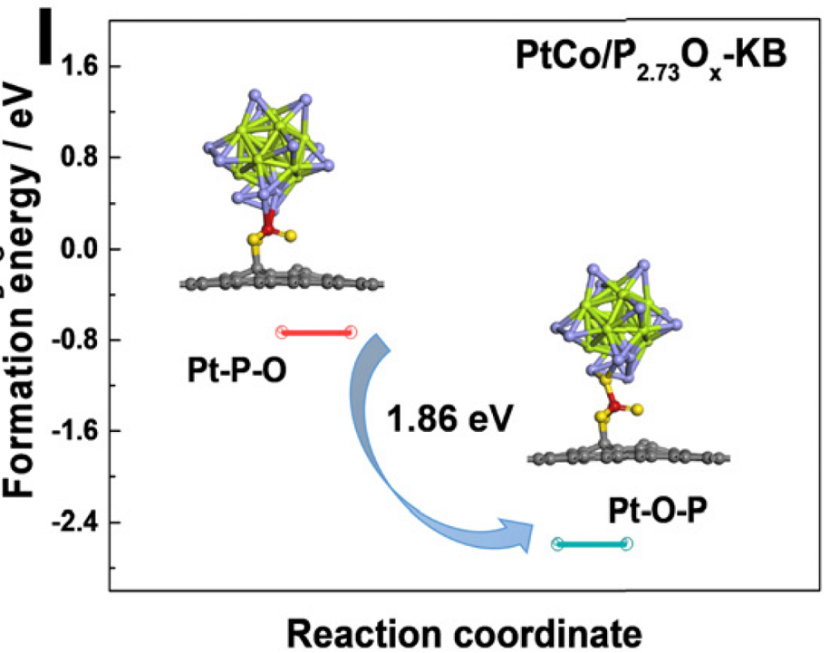
DOI:10.1039/D3EE04503J
总结
在电催化材料设计中,形成能作为核心热力学参数,贯穿催化剂稳定性筛选、活性位点预测及反应路径优化的全流程,为理性设计提供关键判据。
研究者通过密度泛函理论(DFT)计算不同构型(如单原子位点、合金相)的形成能,可快速识别热力学稳定相并排除易失活组分,例如Fe-N₄位点相较于其他配位形式更低的形成能直接预示其作为主要活性中心的优势。
进阶分析中,形成能与电子结构参数(如电荷密度分布、d带中心位置)的耦合计算可深入解析催化活性起源:电荷转移程度决定中间体吸附强度,而d带中心偏移则关联着催化剂的氧化还原能力,二者共同构建了“结构-电子特性-活性”的定量关系网络。
计算实践中,VASP、Quantum ESPRESSO等软件通过优化k点网格与能量收敛标准实现高精度模拟,配合自动化脚本批量处理数据,显著提升计算效率。
这种多尺度理论框架不仅能够预测材料在电化学环境中的动态演化(如表面重构、官能团解离),还可通过构建描述符数据库指导实验合成路径,将传统试错式研发转化为定向设计模式,大幅降低开发成本。
当前研究正通过集成机器学习与高通量计算,进一步拓展形成能分析在多元复合催化剂体系中的应用边界,推动电催化材料设计从经验驱动向数据驱动的范式转变。
找华算做计算?专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
