

说明:原子掺杂是重要的材料调控策略,通过引入杂质原子改变材料电子结构、能带和载流子浓度,从而提升其催化、电学和光学性能。
近年第一性原理计算发展迅速,DFT成为揭示掺杂机理和缺陷态的主要手段。基于DFT提出了多种掺杂策略,如单原子掺杂、共掺杂等,广泛应用于二维材料、半导体、MOFs和金属氧化物体系。
此外,机器学习辅助掺杂预测的方法也逐渐出现,加速了新材料筛选。未来,借助DFT计算指导实验,原子掺杂有望促进高性能多功能材料的发展。



什么是掺杂?
原子掺杂通常以替位或间隙方式将异质原子引入晶格。在半导体和低维材料中,浅层替位掺杂可改变导电类型(实现n型/p型掺杂),而引入过渡金属原子往往在材料中产生局域磁矩,从而引发磁性变化。
DFT计算为研究这些现象提供了关键手段。研究者常通过构建大超胞进行杂质替换掺杂,并计算缺陷形成能来评估掺杂体系的热力学稳定性。Li等基于DFT的缺陷计算为掺杂和缺陷控制提供了重要指导。
这些计算可以定量分析掺杂所引入的缺陷态、载流子浓度及其对电子结构的影响,从而为掺杂策略设计提供理论依据。
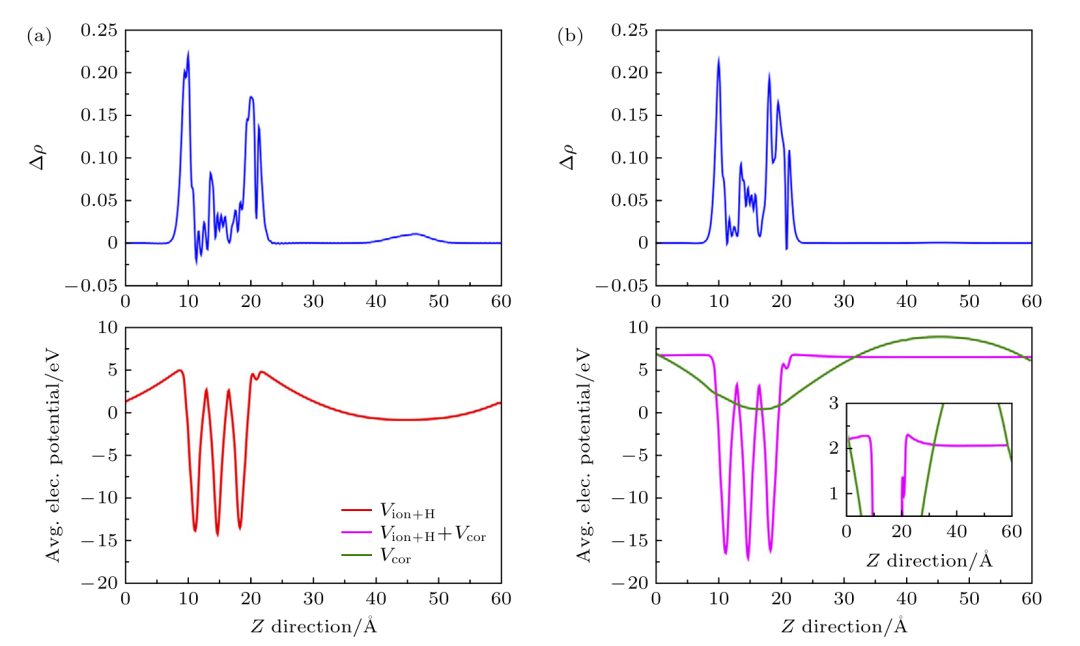
DOI:10.7498/aps.73.20231960
在实际计算中,为克服常规DFT(如GGA/LDA)在带隙和缺陷能级计算上的不足,研究者常引入LDA+U、杂化泛函或带隙校正算符等方案对计算结果进行校正。
此外,为了准确模拟掺杂体系的磁性,需要采用自旋极化计算并分析自旋极化态密度。第一性原理模拟通常结合态密度(DOS)、偏振电荷密度和态分布分析,揭示掺杂原子与主材相互作用的电子本质。
例如,通过对比掺杂前后材料的电子结构,DFT研究可揭示掺杂是否产生中间能级或导带/价带的变化,这对设计半导体掺杂器件尤为重要。
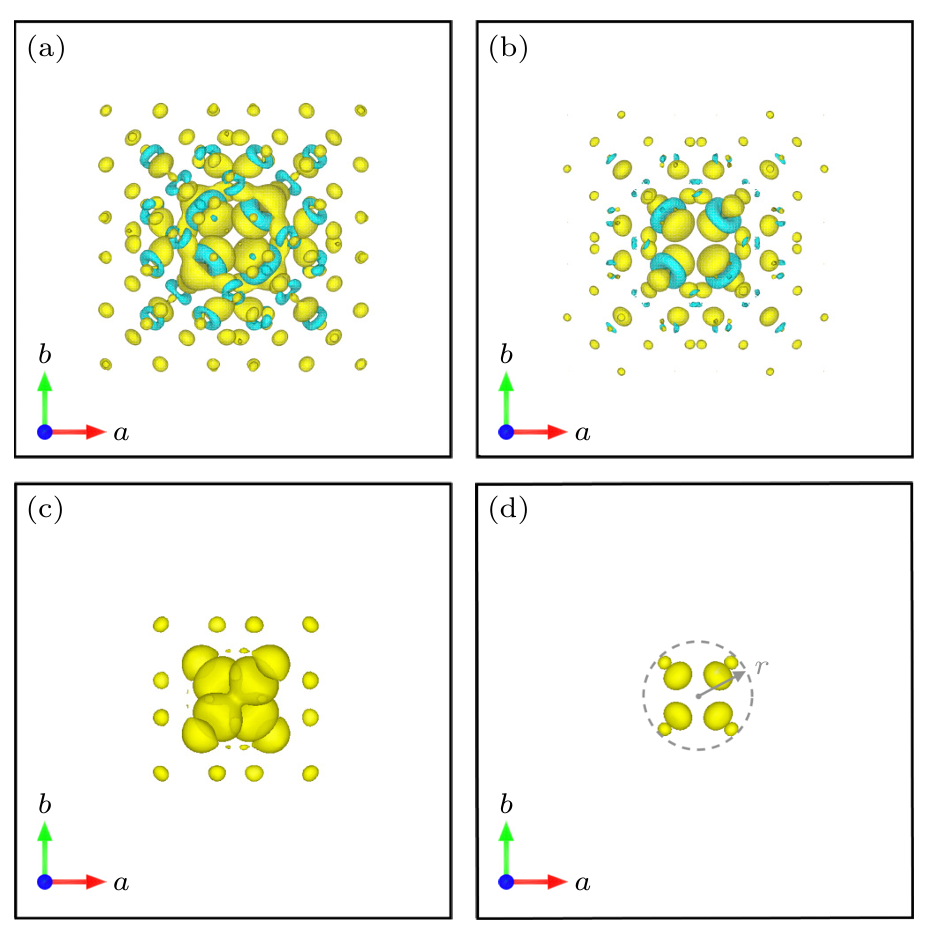
DOI:10.7498/aps.73.20231960
不同体系如何掺杂?
在二维材料和传统半导体中,掺杂是调控性能的常见手段。对于石墨烯等二维材料,DFT研究表明引入N、B、S等杂原子可以显著调制吸附能和导电性,从而改善传感和催化性能。
Peña-Castañeda等基于DFT的研究证实掺杂改性可显著提升石墨烯基有毒气体传感器的灵敏度。
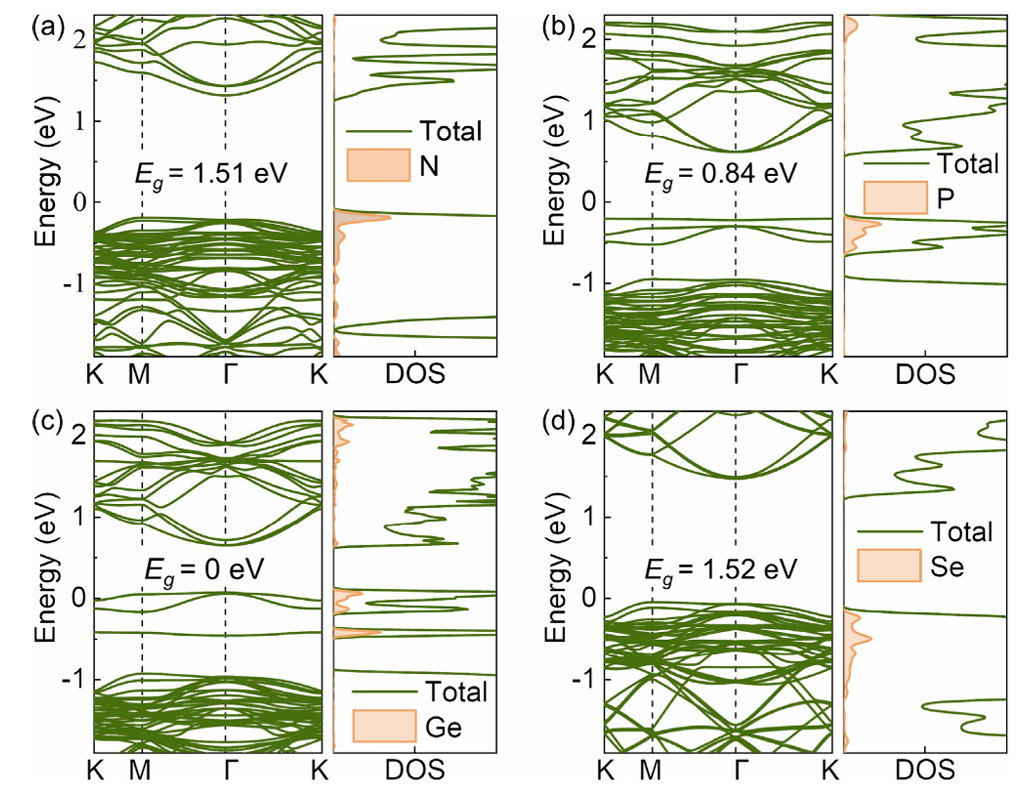
类似地,过渡族二硫化物经过掺杂后也表现出优异性能。例如,Li等通过DFT计算研究了SnS₂单层中N、P、Ge、Se原子的替代掺杂对H₂S吸附的影响,结果显示N和P掺杂显著增强了SnS₂与H₂S的相互作用,同时保持了物理吸附本质;掺杂后SnS₂的带隙分别下降到1.51 eV和0.84 eV,大幅提高了气敏灵敏度。
此外,对传统半导体材料如硅、氮化镓等的掺杂研究也十分丰富,杂原子掺入可以有效调节载流子浓度和迁移率,提高器件的性能和稳定性。
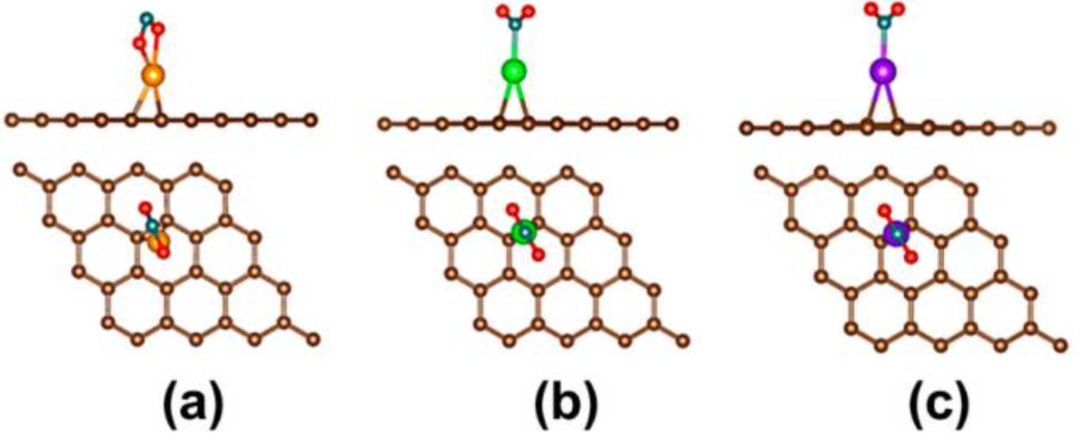
DOI:10.3390/s21061992
对于多孔材料(如MOFs)和金属氧化物,掺杂调控策略也在不断拓展。研究者在金属有机框架中引入不同金属离子或有机配体,以调整孔道性质和催化活性;比如将过渡金属离子掺入ZIF-8等MOF中,可显著改善其对气体分子的吸附能力。
金属氧化物领域中,通过掺杂非金属元素(如N、C)或过渡金属离子来调节其光学吸收和电学性能。例如,掺杂N的TiO₂在可见光光催化方面显示出更高活性;掺杂铝或铜的ZnO可以改变载流子浓度,调整光电检测灵敏度。
这些掺杂策略均得益于DFT计算对结构和电子特征的预测,为实验设计提供方向。
掺杂调控活性与路径
在催化领域,原子掺杂经常被用来构筑活性中心或调节反应路径。例如,在析氢(HER)和析氧(OER)反应中,MoS₂中引入V、Co等过渡金属或杂原子可显著优化其催化活性。
一些研究报告表明,通过在碳材料中掺杂N、P并负载单原子过渡金属,可以制备具有高ORR/OER活性的单原子催化剂。此外,在Li–S电池、电池电极材料中也发现掺杂具有重要作用。
DFT研究揭示,掺杂有助于强化硫化物与电极的结合,降低多硫离子扩散和极化,从而提升循环稳定性和容量。掺杂策略同样应用于纳米硅、LiFePO₄等阴极材料,用于提升离子扩散和电子传导。
在光电子和光催化应用方面,掺杂同样效果显著。例如Tang等向MAPbBr₃钙钛矿纳米晶掺入Mn²⁺可显著提高材料的光致发光效率和结构稳定性。
他们报告Mn掺杂使量子产率在掺杂浓度17%时达到72%,同时抑制了非辐射复合;Mn²⁺填补了晶格中的空位和陷阱态,并促进晶体自组装,提高了晶体完整性。
类似地,在光电探测和光催化领域,通过对g-C₃N₄、TiO₂、ZnO等材料进行掺杂,可以调节其能带结构,提高光吸收和载流子分离效率,进而提升太阳能电池及传感器等器件性能。
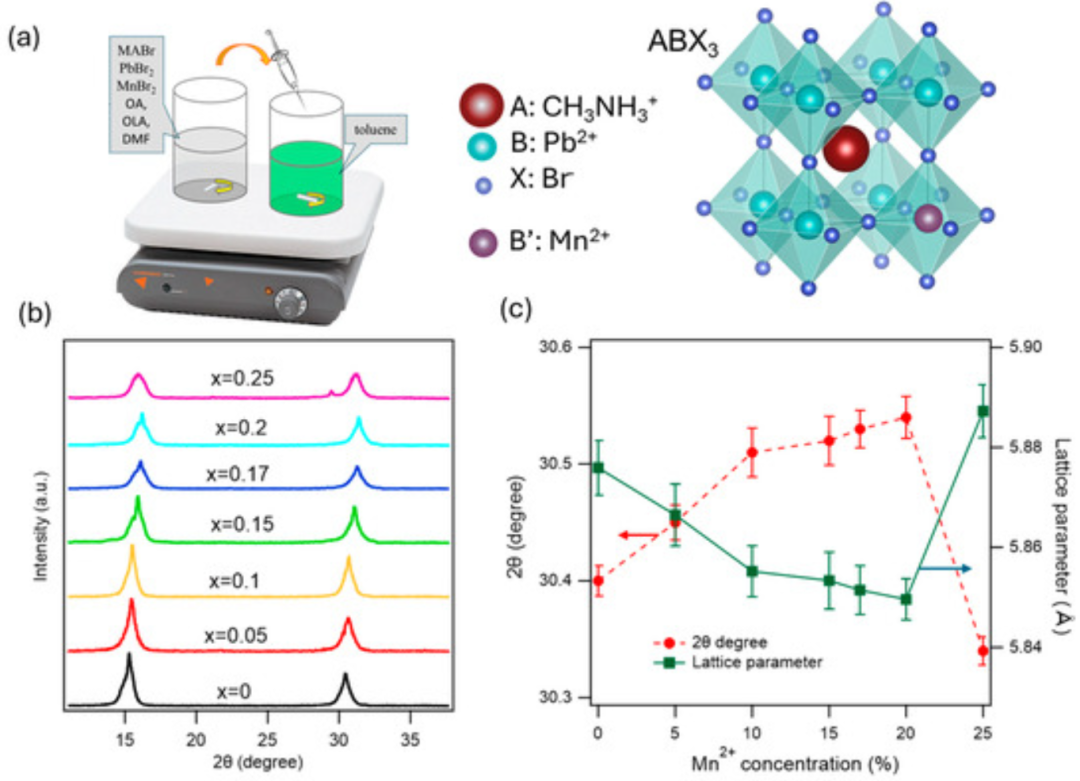
DOI:10.3390/nano15110847
掺杂引起的变化是什么?
DFT计算能够详细揭示掺杂对材料电子结构的影响。一般来说,掺杂会在带隙中引入新的缺陷能级,并改变态密度分布。
正如Li等对于掺杂的SnS₂,N/P掺杂使其带隙分别降至1.51 eV和0.84 eV,生成了额外的杂质态。这些缺陷态对费米能级附近的电子分布产生影响,从而调节材料的电学性质。
许多研究利用部分态密度(PDOS)分析发现,掺杂原子往往贡献显著的杂质DOS峰。
DOI:10.1021/acsanm.4c04339
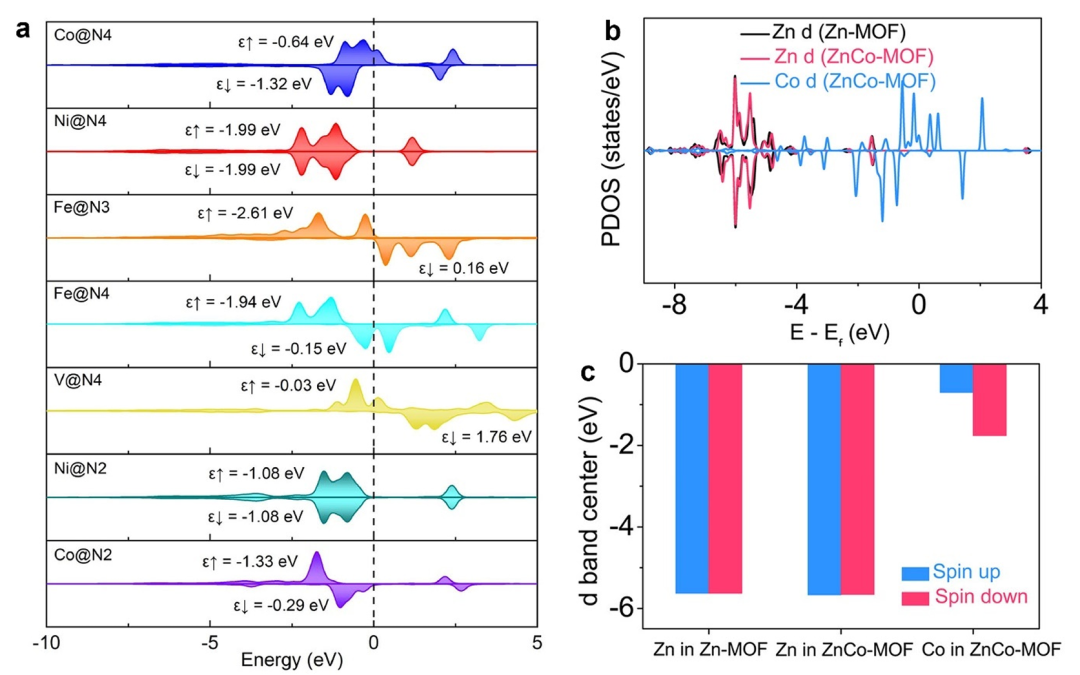
同时,掺杂还会引入或改变缺陷态和局域磁性。例如,向氧化物中掺入磁性元素通常会形成自旋极化的d轨道态,为材料带来磁性。
Chen等将Fe掺入Co₃O₄后,其Fe掺杂体系的d带中心ε_d由原先的-2.01 eV提升至-1.95 eV,这提升了其反键轨道能量,使Fe-Co₃O₄与吸附物的相互作用更加紧密。
DOI:10.3390/molecules28217304
类似地,对掺杂体系的自旋密度分布和电子转移进行分析,可以解释磁性掺杂的机理。通过这些DFT结果,研究者能够量化掺杂对态密度中心、费米能级偏移以及磁矩等指标的影响,从而理解掺杂带来的电子结构变化。
总结
尽管基于DFT的原子掺杂研究取得了诸多成果,但仍面临不少挑战。一方面,传统DFT计算通常基于平衡晶格和有限超胞,难以完全模拟高浓度、多种掺杂原子并存以及实验条件下的热力学不平衡情况。
同时,DFT方法对交换–关联泛函的选择、带隙低估等固有局限依然存在,尽管引入+U和杂化泛函有所改善。另一方面,从计算结果到实验验证往往存在差距:某些理论预测的掺杂配置在实验中难以稳定存在,导致理论指导效果受限。
此外,大规模体系(如纳米结构、缺陷集群)的DFT计算代价高昂,需要更高效的模拟工具。
未来方向方面,DFT与机器学习的结合正成为研究新趋势。有研究将DFTB计算与机器学习相结合,针对不同尺寸和掺杂水平的Zn掺杂MgO纳米粒子开发了加权k近邻模型,用于预测其电子态密度。
这一方法表明,通过机器学习可以大幅加速掺杂体系的电子结构预测。展望未来,结合高通量DFT计算、数据驱动设计和先进实验表征,将有助于系统筛选和发现新型掺杂材料。
此外,多尺度模拟和第一性原理嵌入场景模拟也有望更加真实地再现实验条件,为原子掺杂调控设计提供更精确的理论支撑。