VASP(Vienna Ab initio Simulation Package)是一种广泛应用于材料科学和凝聚态物理领域的第一性原理计算软件。
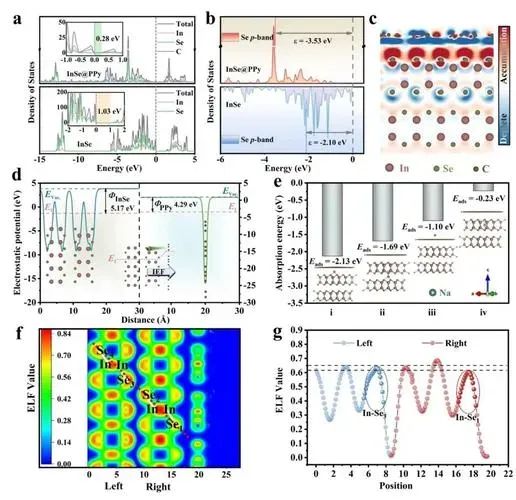
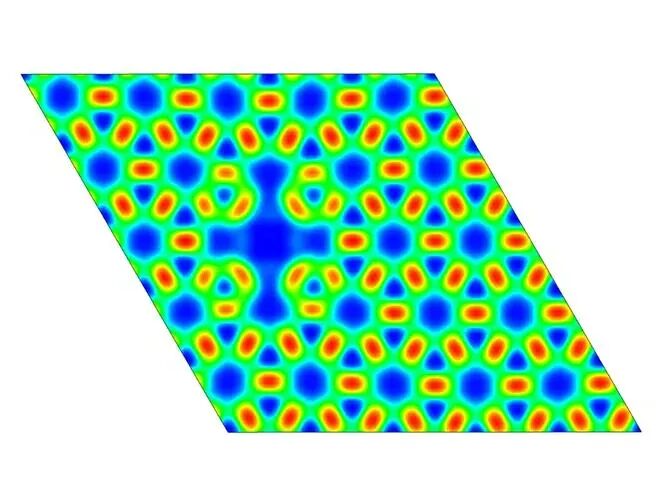
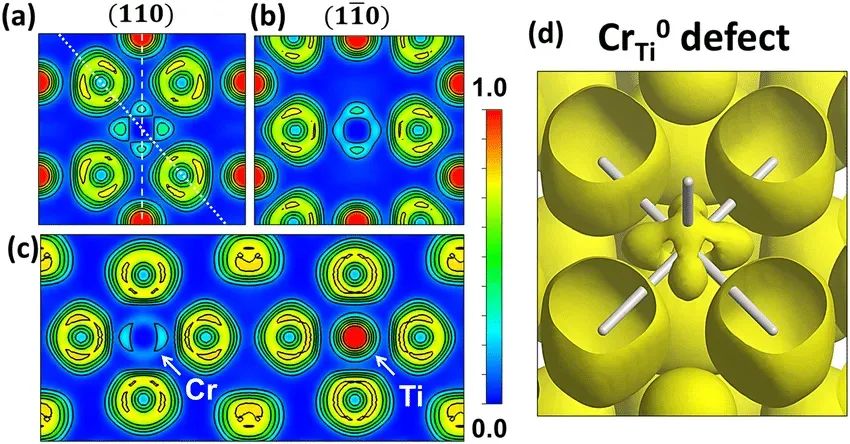
电子局域化函数(Electron Localization Function, ELF)是描述电子在空间中局域化程度的重要工具,其值范围在0到1之间,其中ELF=1表示电子完全局域化,ELF=0表示电子完全离域化。华算科技朱老师通过VASP计算ELF,可以分析材料中的化学键类型、电子结构和成键特征,例如共价键、离子键和金属键的识别。
在进行ELF计算之前,需要对体系进行结构优化,以确保计算的准确性。结构优化通常使用IBRION=2或IBRION=3参数,通过离子弛豫使体系达到能量最低点。
在结构优化完成后,进行自洽计算(SCF)并启用ELF计算,需在INCAR文件中设置LELF=.TRUE.,以启用ELF计算。此外,为了提高计算精度,建议使用高精度模式(PREC=Accurate)和适当的收敛标准(如EDIFF=1E-6)。
计算完成后,生成的ELFCAR文件存储了ELF分布数据。通过VESTA等可视化工具,可以直观地观察电子在不同位置的局域化程度。VESTA支持三维可视化、二维切面图生成和一维数据导出等功能,便于进一步分析ELF的分布情况。例如,通过调整等值面值(isosurface)和晶面取向,可以更清晰地观察电子局域化区域。
在VASP计算ELF过程中,可能会遇到一些常见问题,例如ELFCAR文件未生成、图像不连续区域或定量分析困难等。针对这些问题,可以采取以下解决方案:
检查INCAR文件中是否设置了LELF=.TRUE.,并确保自洽计算的收敛性。
使用VESTA的Line Profile功能沿特定路径提取ELF值,结合其他电子性质(如电荷密度、态密度)进行综合分析。
ELF计算对计算资源有一定要求,建议使用高精度模式(PREC=Accurate)和适当的K点采样。对于大体系,可以调整电荷密度网格(NGXF/Y/Z)和并行计算设置(NPAR=1)以提高计算效率。
ELF计算在材料科学、化学和凝聚态物理领域具有广泛应用。通过ELF分析,可以识别材料中的化学键类型、电子结构和成键特征,例如共价键、离子键和金属键的识别。
此外,ELF还可以用于分析催化活性位点、锂离子电池中Li+迁移通道和材料表面吸附位点的电子特性。
VASP计算电子局域化函数(ELF)是一种强大的工具,能够帮助研究人员深入理解材料的电子结构和成键特征。
通过合理的参数设置、可视化分析和问题解决策略,可以有效提高计算的准确性和效率。随着计算方法的不断进步,ELF在材料科学和凝聚态物理领域的应用前景将更加广阔。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!