

 说明:本文华算科技系统解答了同步辐射技术的15个核心基础问题,涵盖了XAS、XANES、EXAFS的区别与联系、测试模式选择、数据解读与拟合分析等关键内容,为深入理解同步辐射原理与应用、准确获取材料局域结构信息提供清晰实用的指南。
说明:本文华算科技系统解答了同步辐射技术的15个核心基础问题,涵盖了XAS、XANES、EXAFS的区别与联系、测试模式选择、数据解读与拟合分析等关键内容,为深入理解同步辐射原理与应用、准确获取材料局域结构信息提供清晰实用的指南。同步辐射技术是现代科研的重要工具,广泛应用于多个学科领域。然而,若不了解其基础问题,如光源与分析方法的区别、不同光谱技术的联系与差异,以及样品测试和数据处理的关键要点,便难以准确解读实验数据并获取可靠的结构信息。
这些基础问题虽看似简单,实则是掌握同步辐射技术的核心。只有夯实基础,才能深入探索物质的微观结构,推动科学研究的发展。
Q1. 同步辐射与同步辐射XAS有什么区别?
同步辐射是一种带电粒子在磁场中沿曲线运动时发出的电磁辐射,覆盖从红外到X射线的广泛波段。
同步辐射XAS(X射线吸收光谱)是利用同步辐射中的X射线部分,研究物质对X射线的吸收特性,从而分析材料的电子结构和化学状态。
简而言之,同步辐射是一种光源技术,而同步辐射XAS是一种基于该光源的分析方法。
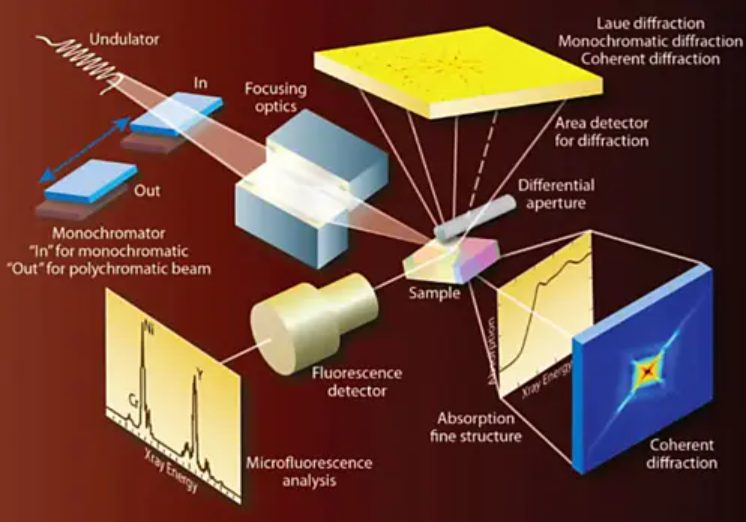
图1:基于同步辐射的表征手段
Q2. XAS与XAFS有什么区别?
同步辐射吸收谱英文全称是X-ray absorption spectroscopy,简称为XAS。XAFS是英文X-ray absorption fine structure的缩写,其中文含义是X射线吸收精细结构。一般来说,XAS与XAFS是一个意思,两种缩写表达。
Q3. XAFS、XANES以及EXAFS有何种关系?
XAFS即X射线吸收精细结构(X-ray Absorption Fine Structure),是指X射线吸收系数在吸收边附近的精细结构。
当X射线照射到物质上时,原子中的电子会吸收X射线的能量并发生跃迁。在吸收边附近,吸收系数随X射线能量的变化不是平滑的,而是呈现出一些振荡结构。
XAFS可分为两个区域,即近边X射线吸收精细结构(XANES,X-ray Absorption Near-Edge Structure)和扩展X射线吸收精细结构(EXAFS,Extended X-ray Absorption Fine Structure)。
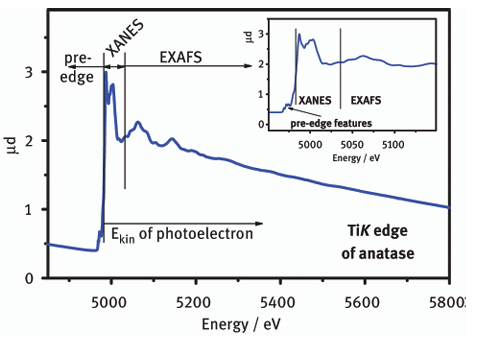
图2:XAS图
Q4. XAS/XAFS能获得哪些信息呢?
XANES区域通常在吸收边附近0-50eV范围内,它对原子的局部几何结构和电子结构非常敏感,能提供诸如原子的氧化态、化学键的类型和对称性等信息。
EXAFS区域一般在吸收边以上50-1000eV甚至更高能量范围,主要反映吸收原子周围近邻原子的种类、数量、距离和无序度等结构信息。
Q5. 一个样测多个元素需要准备几份样品?
如果需要在同步辐射XAS测试中对一个样品中的多个元素进行分析,通常需要准备至少一份样品。因为同步辐射XAS技术可以在同一份样品上依次测量不同元素的吸收光谱,无需将样品分成多份。
不过,如果样品的均匀性较差,或者需要进行多次重复测量以确保数据的可靠性,可能需要准备额外的样品。
Q6. 测试样品应该选荧光模式还是透射模式?
选择同步辐射XAS的透射模式还是荧光模式,主要依据以下几点:
1、元素含量:高含量元素适合透射模式,因为信号强、分析直接;低含量或痕量元素则优先选择荧光模式,因其灵敏度高,能有效捕捉弱信号。
2、样品厚度和吸收特性:薄样品或低吸收体系适合透射模式;厚样品或强吸收体系则更适合荧光模式,以减少信号衰减和失真。
3、研究目标:若需获取结构信息(如晶格参数、密度分布),透射模式更合适;若侧重于元素种类和分布分析,荧光模式是更好的选择。
4、元素属性:对于重元素(高Z值),透射信号更稳定;而对于轻元素(低Z值),荧光信号更易区分,优先选择荧光模式。
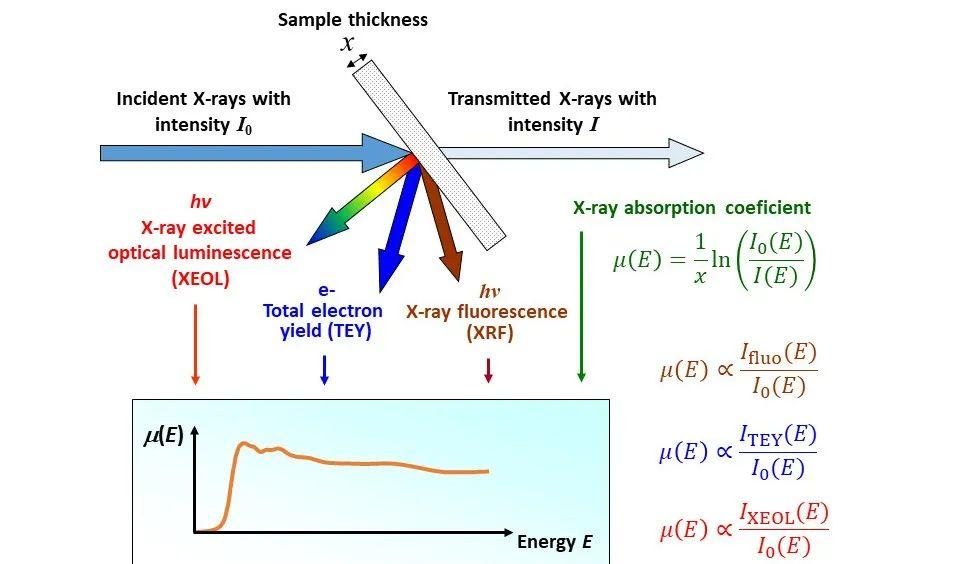
图3:X射线与物质相互作用的原理图
Q7. 测试同步辐射如何选用标样
一般定性不需要测,定量需要测试。选择同步辐射测试的标样时,需确保标样的化学组成、晶体结构和电子状态与待测样品尽可能接近,以保证数据的准确性和可比性。
Q8. E空间、K空间、R空间各能得到什么信息?
E空间:能量空间,主要反映吸收边的位置、形状和强度等信息。通过观察吸收边的跳变高度、近边结构和扩展边的曲线特征,可以获得样品的电子结构信息。然而,由于样品浓度、X射线光强和采集模式等因素的影响,不同条件下测得的吸收边高度会有所不同。
为了消除这些因素的干扰,使数据具有可比性,需要对实验数据进行归一化处理。归一化处理是将XAFS谱的吸收边强度标准化为“1”,从而确保不同实验条件下的数据能够在同一标准下进行比较。
K空间:波矢空间,通过对E空间的XAFS数据进行转换得到。将数据从能量空间转换到波矢空间的目的是为了得到等间隔的k值,以便进行后续的傅里叶变换。
在XAFS数据采集过程中,高能部分的信号通常会显著衰减。因此,在转换到K空间后,高k值部分的信号也会出现衰减。
为了恢复这部分信号的权重,通常会对数据进行加权处理,即使用knc(k)的形式,其中n一般取1、2或3。加权的目的是为了更好地恢复高k值部分的信号,从而更准确地反映样品的真实结构。
R空间:通过傅里叶变换将加权后的K空间数据转换得到的径向分布函数。R空间曲线中的不同峰代表了不同位置的配位原子。
通过分析R空间图,可以直观地获取配位原子的信息,例如峰的位置反映了配位原子的键长,峰的强度则与配位原子的数量和无序度有关。这些信息对于理解样品的局部结构和化学环境非常关键。
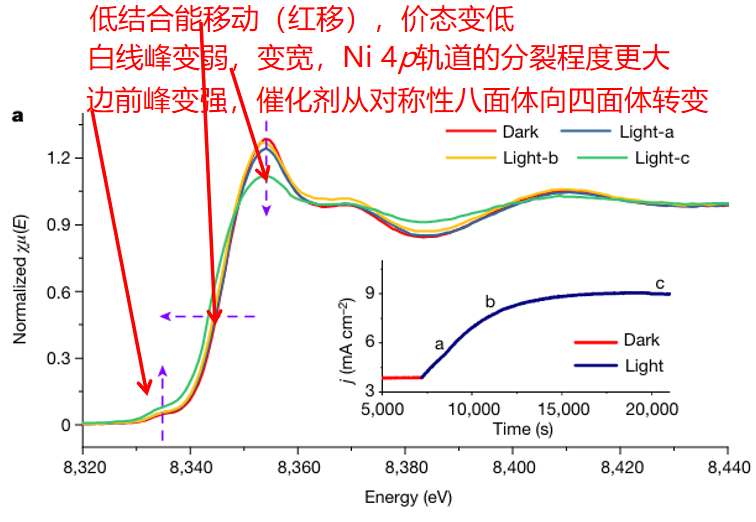
图4:E空间、K空间、R空间转换
Q9. E空间中的XANES如何确定价态和对称性等信息?
价态的确定:
吸收边位置(结合能):吸收边的位置(即X射线吸收边的起始位置)是判断元素价态的重要指标。吸收边位置越高,表明电子从内层跃迁到更高能级所需的能量越大,通常意味着更高的价态。
边前峰(Pre-edge peaks):边前峰是指吸收边之前的小峰,这些峰通常与未占据的d轨道有关。边前峰的存在和强度可以提供关于价态的额外信息。
对称性的判断:
边前峰的对称性:边前峰的形状和对称性可以反映样品的对称性。如果边前峰是对称的,可能表明样品具有较高的对称性;如果不对称,则可能表明样品的对称性较低或存在无序。
白线峰(White line):白线峰是指吸收边之后的一个显著的峰,其位置和强度可以反映轨道的分裂程度。白线峰的形状和位置变化可以提供关于样品对称性的信息。
轨道分裂程度:
白线峰的强度和位置:白线峰的强度和位置可以反映轨道的分裂程度。在晶体场中,d轨道的分裂程度会影响白线峰的强度和位置。
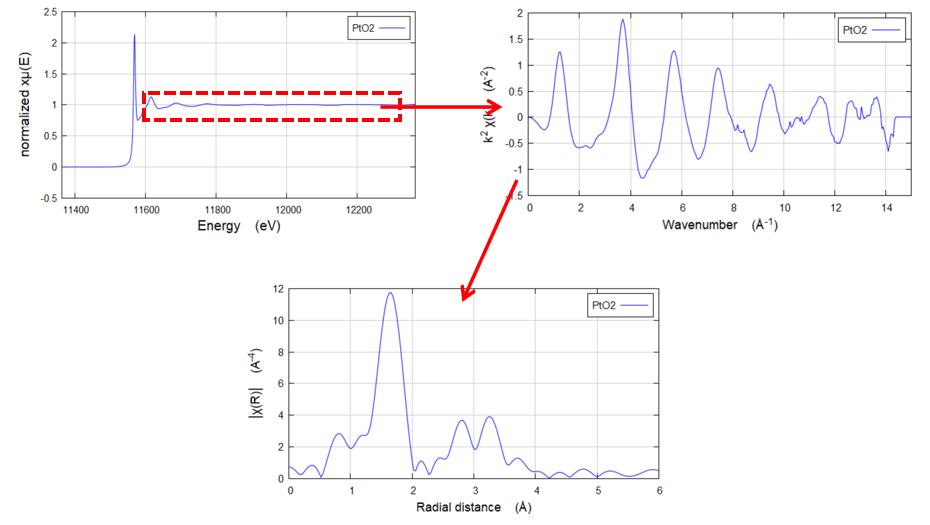
图5:NR-NiOOH在接受光照后的Ni-K边XANES光谱
Q10. E空间中的EXAFS能知道什么,为什么需要拟合?
在E空间中,EXAFS提供了吸收原子周围近邻原子的结构信息,包括配位数、键长和无序度。具体如下:
1、配位数
振幅分析:EXAFS谱振幅与配位数成正比,振幅越大,散射强度越强,配位数越高。
拟合分析:拟合EXAFS数据时,配位数是关键参数,优化拟合可得准确配位数。
2、键长
相位分析:EXAFS谱相位变化与键长相关,相位变化反映路径长度,可推断键长。
拟合分析:拟合时键长是重要参数,优化拟合可得准确键长。
3、无序度(Debye-Waller因子)
振幅衰减:EXAFS谱振幅衰减与无序度相关,无序度越高,衰减越明显。
拟合分析:拟合时无序度是关键参数,优化拟合可得准确无序度。
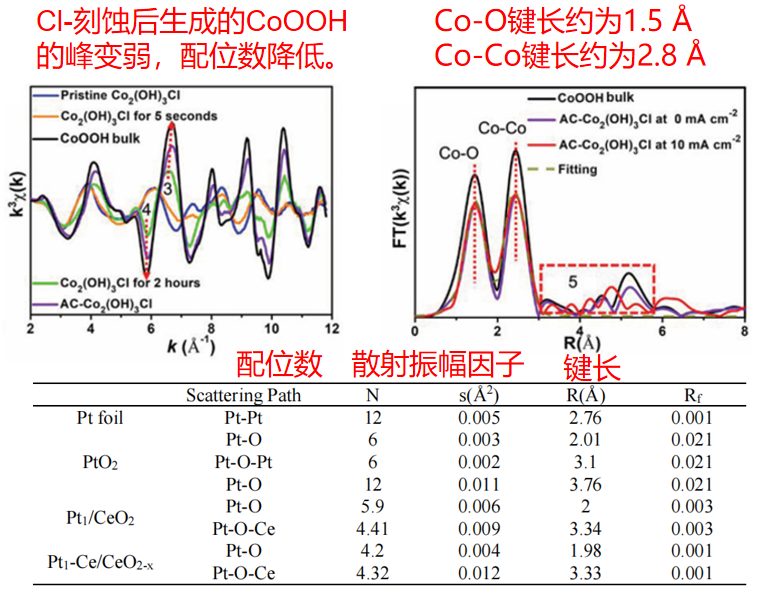
图6:K空间、R空间、拟合
Q11. 为什么给出的R空间谱图中的键长与拟合参数表中的键长不一样呢?
在R空间谱图中,峰的位置并不代表吸收原子与配位原子之间的实际键长。通常情况下,实际键长比R空间图中峰的位置要长0.3~0.5 Å左右,具体差异会因吸收原子的种类而有所不同。
为了获得更准确的键长信息,通常需要利用Atermis软件对EXAFS(扩展X射线吸收精细结构)数据的壳层部分进行拟合分析。拟合得到的所有参数,包括精确的键长,都会详细列在拟合参数表中。
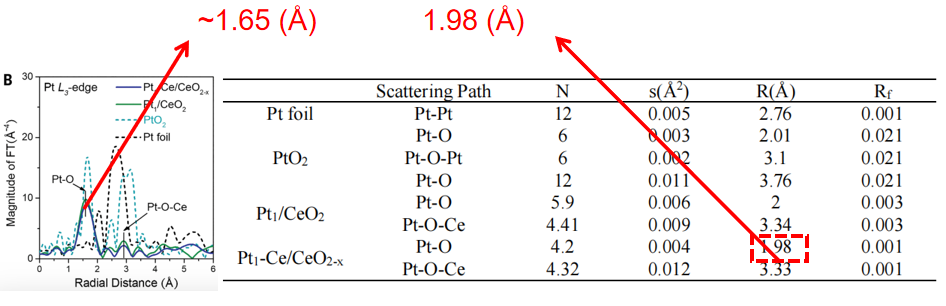
图7:R空间谱图中的键长与拟合参数对比
Q12. R空间谱图中不同峰的位置是否有特征性呢?
R空间谱图中的峰位置本身并无绝对特征性,本质上是傅里叶变换后的数学曲线,需结合具体样品信息和实验条件分析。这些峰可能是多种元素峰的叠加,需通过分峰拟合来判断各峰的来源和贡献。
同一种吸收元素在R空间图中的峰位置也不一定代表相同的配位结构,因为峰的位置受到键长、配位数、无序度和相位因子等四个参数的共同影响。仅用配位数来解释峰的位置是不严谨的。
在拟合时,可以通过固定其他三个参数(如键长、无序度和相位因子)来对比配位数,从而获得更可靠的结构信息。
此外,R空间图中3 Å以后的峰往往更多地受到噪声和高能量衰减的影响,其结构信息含量较低,很多峰并无明确物理意义。因此,分析R空间谱图时,需综合考虑多种因素,谨慎对待每个峰的物理意义。
Q13. 拟合参数表中提供的配位数与材料实际的结构是什么关系呢?
通过Atermis软件对EXAFS谱图进行拟合后,得到的是样品中吸收原子整体平均配位的数量,并不能像单晶材料那样可以得到具体的晶胞参数等信息,有的是以吸收原子为中心,向外不断延伸至其近邻的原子之间平均配位情况。
并且这种结构关系仅仅是配位数量,与其键角以及对称性等是没有太大关系的,所以我们通常得到的配位数是无法判断其是平面还是立体的,只有对材料有预设建模,并且多种检测共同能够验证其结构状态,才可以系统性的对比哦。
Q14. Wavelet小波变换能得到哪些信息呢?
小波变换是一种强大的数据分析工具,能够对XAFS(X射线吸收精细结构)数据进行高分辨率分析。它通过在二维或三维空间中展开K空间和R空间数据,提供关于样品局部结构的详细信息。
在二维小波变换图中,不同颜色代表峰的高度,对应R空间图中峰的强度,可以直观地区分配位原子的距离和种类。
小波变换特别适用于区分相邻键长相近的键以及解析复杂结构中的不同配位环境,尽管主要用于定性分析,但它为后续的定量分析提供了重要的线索和基础。
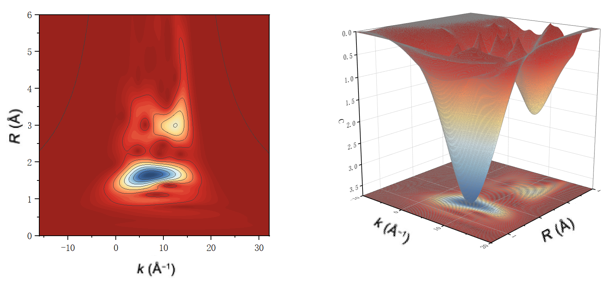
图8:小波变换图
Q15. 怎么样通过吸收谱来判断我做的材料是不是单原子呢?
在进行单原子材料的吸收谱测试前,建议先通过球差电镜观察其是否具有单原子分散结构。根据我们的经验,如果电镜图像中出现大量单分散亮点,吸收谱测试通常能得到与之匹配的结果。
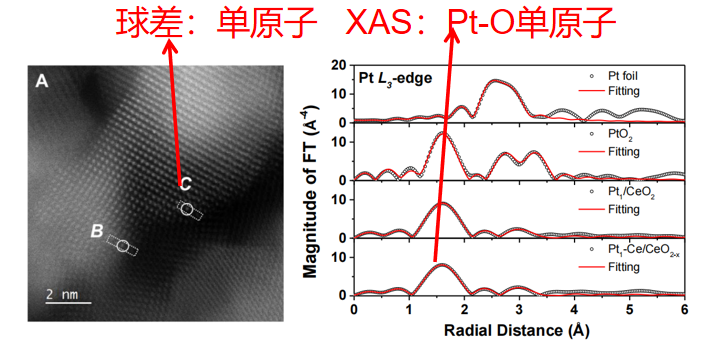
图9:单原子表征
Q16、17……大家还有什么问题呢?