

 氧析出反应(Oxygen Evolution Reaction, OER)是水分解制氢过程中动力学最缓慢的半反应,需要同时转移四个电子才能将水氧化为氧气。
氧析出反应(Oxygen Evolution Reaction, OER)是水分解制氢过程中动力学最缓慢的半反应,需要同时转移四个电子才能将水氧化为氧气。OER在酸性和碱性电解质中均可发生,但两种介质下的反应机理存在显著差异,尤其在中间体物种、质子/氢氧根参与方式以及可能的“吸附质演化机制”(Adsorbate Evolution Mechanism, AEM)和“晶格氧演化机制”(Lattice Oxygen Mechanism, LOM)的贡献方面。
理解酸碱条件OER机理差异对于设计高效电催化剂具有重要意义。本文华算科技围绕典型非贵金属OER催化剂NiFe层状双氢氧化物(NiFe-LDH),系统综述酸性OER与碱性OER的反应路径差异,介绍自由能台阶图的构建方法及其判断速率控制步骤和活性的方法,并解析理论计算中常用的各种OER数据图(ΔG中间体自由能图、塔菲尔图、总态/投影态电子密度DOS/PDOS、差分电荷密度图等)的含义和解读方法。
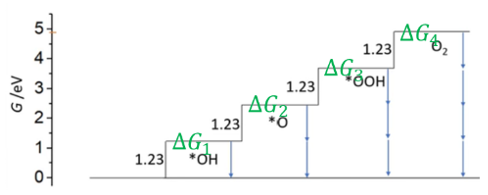
理想台阶图
酸性与碱性OER路径及中间体差异
酸性介质中的OER机理:在酸性条件下,OER以水为反应物,典型机理包括四个逐步的质子-电子转移步骤。常见路径为:表面活性位上的金属中心(记为M)首先吸附一分子水并脱去一质子,形成吸附的羟基中间体OH,同时释放一个质子和一个电子(M+H₂O→M–OH+H⁺+e⁻)。
接着,OH中间体进一步脱氢,产生吸附的氧原子O,并再释放一对H⁺/e⁻(M–OH→M–O+H⁺+e⁻)。第三步,另一个水分子进攻O,形成过氧羟基中间体OOH,同时释放H⁺/e⁻(M–O + H₂O → M–OOH + H⁺ + e⁻)。
最后,OOH脱去最后一个质子和电子生成气态O₂,催化剂表面恢复初始状态(M–OOH→M+O₂↑+H⁺+e⁻)。
在该“吸附质演化机制”(AEM)中,每个步骤均涉及质子和电子的协同转移,即质子从水中脱出并随电子一起转移到电极。酸性环境中典型的中间体就是上述OH、O、*OOH三种吸附物种。
碱性介质中的OER机理:在碱性条件(如氢氧化钾溶液,pH≈14)下,OER以氢氧根离子 (OH⁻) 为质子提供体,其机理与酸性类似但质子来源和产物不同。碱性OER的一般路径为:首先,OH⁻吸附在催化剂金属位形成OH,释放一个电子(M+OH⁻→M–OH+e⁻)。
随后,OH被另一OH⁻抽氢生成吸附氧O,并产出一分子水和电子(M–OH+OH⁻→M–O+H₂O+e⁻)。第三步,O与溶液中的OH⁻发生亲核反应生成OOH,中间伴随电子转移(M–O+OH⁻→M–OOH+e⁻)。
最后,OOH被OH⁻去质子形成O₂并释出,表面位点重新生成OH完成循环(M-OOH+OH⁻→M–OH+O₂↑+e⁻)。可以看出,碱性介质下每步反应消耗OH⁻且释放电子,同时产生水而无自由质子累积。
酸性与碱性路径在吸附中间体种类上都是 OH、O、OOH,但质子在体系中的角色截然不同:酸性下质子作为产物被不断移走,碱性下则通过OH⁻提供质子并生成水。此外,电催化表面在碱性条件往往天然被–OH覆盖,初始状态常是M–OH(而非裸金属),因此有时将第一步视为 *OH的进一步脱氢过程。
总的来说,两种介质的基元步骤在本质上类似,但酸性OER通过质子释放平衡电荷,而碱性OER通过OH⁻供给质子并生成水来实现电荷平衡。

AEM与LOM机制的差异:值得注意的是,在不同催化剂和条件下,OER可能遵循上述传统的AEM机制,亦可能涉及晶格氧参与机制(LOM)。AEM机制下氧来自于吸附的水/羟基,步骤都伴随质子-电子协同转移,而LOM机制中,氧的一部分来自催化剂晶格本身。
LOM一般发生在某些过渡金属氧化物/氢氧化物催化剂中,表现为高价金属可以抽取自身晶格氧参与O–O键形成。在LOM路径中,通常催化剂表面的–OH基团经多步失去质子转化为高价的=O或“O⁻”形式(即“晶格氧”被活化),然后两个相邻晶格氧(或一个晶格氧与一个吸附氧物种)直接发生O–O耦合生成O₂,这一步不需要额外电子转移,属于非电化学的化学脱附过程。
随后催化剂晶格中留下的氧空位会被溶液中的OH⁻重新填补,以完成催化循环。这种LOM机制打破了每步都需质子-电子同步转移的限制,被认为可绕过某些AEM路径中能垒较高的步骤,从而提高活性。
然而,LOM往往要求催化剂能够形成并稳定高价态(金属–氧键较弱、氧空位易形成)。酸性环境一般更倾向于AEM机制,因为大多数催化剂在酸中不宜形成氧空位且晶格氧一旦流失易引发结构溶解。
但在碱性环境下(尤其是NiFe-LDH等金属氢氧化物催化剂中),研究发现LOM机制可能占重要地位。例如,NiFe-LDH在高氧化电位下能够发生晶格氧的氧化和再生,即通过LOM路径提升OER活性。
OER自由能台阶图的构建方法
自由能台阶图的构建:自由能台阶图(又称Gibbs自由能梯阶图)是分析OER机理和活性的重要理论工具。其构建基于密度泛函理论(DFT)计算得到的各中间体吸附自由能。
典型做法是采用诺斯科夫等提出的“计算氢电极”(Computational Hydrogen Electrode, CHE)方法,以标准氢电极平衡条件作为参考来计算各步反应的自由能变ΔG。具体而言,在理论上将每一步的质子+电子转移等效为½H₂(g)的生成/消耗,从而避免直接处理显式的带电体系。
计算步骤包括:首先,在模型催化剂表面的活性位上优化吸附各种中间体(OH, O, OOH)的构型并计算其吸附能;然后引入零点能和熵校正得到自由能;最后,将这些自由能值按照反应进程排列绘制成台阶状的能量图。
横轴表示反应坐标依次经历4个基元步骤,纵轴为每一步后的相对自由能。通常在标准平衡电位1.23V(RHE)下考察各步ΔG:理想催化剂在1.23V应使4个步骤的ΔG都为零,从而无需过电位即可驱动反应。
但实际催化剂由于中间体键强的限制,往往无法同时满足OH、O、OOH的最佳吸附能,必然存在某一步ΔG为正值,即为潜在决定步骤(Potential-Determining Step, PDS)。
该步对应的自由能壁垒即表示需要施加的理论过电位η (η=最大ΔG/e–1.23V)。举例来说,许多过渡金属氧化物催化剂存在OH和OOH吸附能的线性关联,导致两者能量差约为3.2eV。
这使得至少有一步ΔG≈3.2eV/每O_2,即需要约0.37V的最低过电位,这被认为是传统AEM路径下OER的活性上限。如果催化剂能打破这种限制(例如通过LOM路径),则有望降低最大自由能垫高,从而降低过电位。
判断速率控制步骤和活性:自由能台阶图直观揭示了各步反应的热力学难易程度。速率控制步骤(RDS)通常对应图中自由能攀升最高的那个步骤。在1.23V条件下,若某一步ΔG远高于其它步骤,则该步骤反应最难发生,因而限制总体反应速率。
例如,在许多催化剂上,OH脱氢为O是最困难的一步,因此被视为RDS,实验上也常通过分析塔菲尔斜率或反应级数验证这一判断(见下节塔菲尔分析)。
另一方面,自由能图还能用于比较不同催化剂的理论活性:最大ΔG越小,理论所需过电位越低,催化剂活性越高。研究者常通过调整催化剂组成和结构来改变中间体的ΔG,从而优化自由能分布使各步ΔG更加平衡。
这种思路在理论上对应火山曲线的概念:过弱或过强的中间体吸附都会导致某步ΔG过大,只有达到适中吸附强度才能接近理想自由能分布。因此,自由能台阶图不仅帮助确定哪一步是限速步骤,也提供了改进催化剂的方向——即针对限速步骤调整相关中间体的键合强度。
需要强调的是,自由能分析反映的是反应热力学驱动力,假设各步均无较高的动力学屏障。实际反应中还需考虑各步的活化能,但一般自由能最高的步骤往往也对应最高的动力学障碍,因此自由能图对RDS的判断在大多数情况下是合理的。
DOI:10.1002/adma.202209307
典型OER数据图的构成与解读
理论研究OER机理和催化活性,常结合多种数据图表以从不同角度揭示关键信息。以下列举典型图像类型及其含义:
ΔG中间体自由能图:如上节所述,这是将4个OER基元步骤的吉布斯自由能变化绘制而成的柱状或折线图。每个台阶高度代表在某一给定电位下,从前一中间体转变为下一中间体的自由能变ΔG。
通过该图可以直接看出哪个步骤ΔG最大(限速步骤)以及该催化剂在该电位下的理论过电位是多少。
DOI: 10.1126/science.adk9849
电子态密度(DOS)与投影态密度(PDOS)图:DOS描述了材料中电子能级的分布情况,即每个能量下存在多少电子态;PDOS则将总态密度投影到特定的原子或轨道,以解析不同元素或轨道对电子态的贡献。
对于电催化剂而言,DOS/PDOS图是理解电子结构-催化性能关系的关键工具。例如,过渡金属氧化物催化剂的金属d带与氧2p带的位置关系被认为决定了M–O键强度,从而影响OER中间体的吸附能。
在PDOS图中,研究者常关注以下特征:(i)d带中心:过渡金属d轨道态密度的重心位置。d带中心较高(接近费米能级)通常意味着金属与氧吸附物的键合较强,而d带中心较低则键合较弱。
优化的d带中心可使中间体吸附强度适中,从而提高OER活性。(ii)O 2p带中心:氧2p态密度重心相对于费米能级的位置。
特别在晶格氧机制中,O2p能带位置被用作氧活性的判据:O2p能级越接近费米能级,意味着氧的价带电子更容易被抽出,晶格氧越容易被氧化参与O₂生成。例如,有研究指出,O2p带中心上移(更接近费米能级)会使晶格氧易于失去电子形成氧空位,即促进LOM发生。
(iii)反键态占据情况:通过PDOS可以观察金属–氧键的反键轨道是否在费米能级下被占据。反键态占据多表明金属–氧键中的电子已填充到不利于键合的轨道,暗示键强下降、更易断裂。
例如,NiFe-LDH中掺入某些杂原子(如Mo)后,发现Ni3d与O2p轨道杂化增强,在费米能级附近的反键态占据增多,导致Ni–O键变得更弱,更易形成氧空位,实现晶格氧参与。
总之,通过DOS/PDOS分析,可以定性了解电子结构的变化如何影响中间体吸附及反应路径。
例如,当发现掺杂使金属d与氧p的态密度重叠增加、反键态填充提高时,往往预示晶格氧活性提高,有利于LOM;而若d带中心明显下移,则可能导致吸附物过弱,活性下降。这些电子结构指标为理性设计催化剂提供了指导。
差分电荷密度图(Charge Density Difference, CDD):这是用来可视化反应过程中电子重新分布的图像。差分电荷密度通常定义为体系在发生相互作用后的电荷密度减去各组分(如吸附前的催化剂表面和气相中间体)的电荷密度之和,即Δρ=ρ(催化剂+吸附物)– ρ(催化剂裸表面)– ρ(吸附物气相)。
通过绘制Δρ的等值面或截面图,可以直观地看到电子在哪些区域发生了累积或缺失。图中常用两种颜色(例如黄色表示电子积累,青色表示电子亏缺)来标识相对于非相互作用状态的电荷变化。
解读方法:差分电荷密度图可以告诉我们键形成或断裂时的电荷转移方向和程度。例如,在OH吸附在金属位时,差分电荷密度可能显示氧原子周围出现电子积累(来自金属提供的价电子),而金属原子与氧之间区域呈电子亏缺,表示金属向氧提供电子形成共价键。
结合定量的Bader电荷分析,可以进一步获得电子转移的数值大小,但差分电荷密度图本身已足以提供反应过程中电子行为的图像化理解。
DFT理论计算获取OER
计算方法与软件:理论上获取上述自由能和电子结构信息,主要依赖于密度泛函理论(DFT)计算。常用的软件包括VASP、QuantumESPRESSO、Gaussian(配合嵌入模型)、CP2K等。
其中VASP因擅长周期性体系的平面波模拟,被广泛用于固体催化剂表面吸附和反应能垒计算。一般流程是:首先构建催化剂的表面模型,如NiFe-LDH常用其最稳定的晶面构建有限层数的剖面(比如(001)晶面)并在两侧加真空层以模拟表/界面。
然后选择合适的交换相关泛函(常用泛函如PBE),针对过渡金属含量高的体系往往还需引入DFT+U方法校正d电子的自相互作用(例如Ni和Fe的3d轨道通常施加一个U参数以更准确描述Ni^3+/Fe^3+高自旋态)。
接着在所选表面活性位上吸附中间体(OH, O, OOH等),对吸附构型进行几何优化以找到最低能量构型。同时考虑自旋极化计算,因为Ni、Fe等过渡金属可能具有未成对电子,强磁性态会影响吸附能和电子结构。
对于NiFe-LDH这种含Fe^3+的体系,初始可以给Fe设定高自旋态,以模拟实验观察到的Fe在NiOOH中的低自旋/高自旋转变行为。
模型构建要点:构建合理的计算模型对可信结果至关重要。以NiFe-LDH为例,它在静态条件下为层状双氢氧化物,中间插有阴离子(如NO₃⁻或CO₃²⁻)平衡电荷。然而在强氧化电位下,NiFe-LDH实际转变为Ni、Fe氧羟化物活性相(类似于NiOOH/FeOOH)。
因此理论计算时通常直接采用NiFeOOH晶格模型:比如以β-NiOOH的层状结构为基底,在Ni位替换一定比例的Fe,从而构造Ni:Fe约3:1(25%Fe)的模型来代表NiFe-LDH的活性相。这样的模型捕捉了Fe掺杂对Ni氧化物的电子结构影响,同时简化了插层阴离子的影响(假定在高电位下阴离子已脱出或作用可忽略)。
模型中需确保整体电荷中性(通常每引入一个Fe^3+取代Ni^2+,需在层间加一额外OH⁻或调整总体H含量)。此外,模型尺寸要足够大以避免吸附物之间的镜像干扰;通常采用2×2或更大的超胞,并在垂直方向加>15Å真空。
溶剂效应也是一大考虑因素:纯DFT为零温真空中的能量,需要对水溶液环境下自由能进行校正,例如加上中间体的溶剂化能近似或采用连续介质模型(如VASP-Sol等)模拟水的介电屏蔽。
自由能计算与过电位评估:在获得吸附态的DFT总能量后,还需考虑零点振动能(ZPE)和熵(S)修正以得到室温下的Gibbs自由能:G = E_DFT + ZPE – T·S。其中ZPE和S可通过计算吸附物和气相物种的振动频率来估算(通常小分子可以直接计算频率,大体系可近似各局部振动模式)。
特别地,气相H₂的G(T)可由热力学数据获得。在酸性条件下,质子-电子对以½H₂参考,则在标准条件(298K,pH0)下½H₂的G = 0,因此各步ΔG = ΔE_DFT + ΔZPE – TΔS – eU(其中eU项表示施加电位U对每转移1e⁻的自由能降低)。
通过在U=1.23V下计算ΔG,可以判断是否有步骤仍为正。如果有,说明该催化剂需要额外过电位η使该步变为平衡,否则此正的ΔG即为导致过电位的热力学瓶颈。这样的分析重复于不同材料,可预测其相对活性高低。
DOI:10.1002/smll.202304260
总结
通过以上分析,我们了解到酸性和碱性条件下OER机理的差异源于质子参与方式和催化剂电子结构的不同。酸性介质中反应遵循质子-电子协同的吸附质演化路径,而碱性介质中OH⁻的参与引出更复杂的机制,尤其是在NiFe-LDH等催化剂中可能出现晶格氧直接参与产氧的LOM路径。
自由能台阶图是理论研究OER的重要工具,它量化了各步反应的热力学要求,可用于判定限速步骤和比较不同催化剂的活性。
事实证明,深入的理论计算与图像分析在电催化研究中已成为不可或缺的一环,它为理解反应本质和催化剂改性提供了清晰的“显微镜”,也为我们走向理性能源催化设计时代奠定了基础。

点击阅读原文,立即下单!?

理想台阶图
酸性与碱性OER路径及中间体差异
酸性介质中的OER机理:在酸性条件下,OER以水为反应物,典型机理包括四个逐步的质子-电子转移步骤。常见路径为:表面活性位上的金属中心(记为M)首先吸附一分子水并脱去一质子,形成吸附的羟基中间体OH,同时释放一个质子和一个电子(M+H₂O→M–OH+H⁺+e⁻)。
接着,OH中间体进一步脱氢,产生吸附的氧原子O,并再释放一对H⁺/e⁻(M–OH→M–O+H⁺+e⁻)。第三步,另一个水分子进攻O,形成过氧羟基中间体OOH,同时释放H⁺/e⁻(M–O + H₂O → M–OOH + H⁺ + e⁻)。
最后,OOH脱去最后一个质子和电子生成气态O₂,催化剂表面恢复初始状态(M–OOH→M+O₂↑+H⁺+e⁻)。
在该“吸附质演化机制”(AEM)中,每个步骤均涉及质子和电子的协同转移,即质子从水中脱出并随电子一起转移到电极。酸性环境中典型的中间体就是上述OH、O、*OOH三种吸附物种。
碱性介质中的OER机理:在碱性条件(如氢氧化钾溶液,pH≈14)下,OER以氢氧根离子 (OH⁻) 为质子提供体,其机理与酸性类似但质子来源和产物不同。碱性OER的一般路径为:首先,OH⁻吸附在催化剂金属位形成OH,释放一个电子(M+OH⁻→M–OH+e⁻)。
随后,OH被另一OH⁻抽氢生成吸附氧O,并产出一分子水和电子(M–OH+OH⁻→M–O+H₂O+e⁻)。第三步,O与溶液中的OH⁻发生亲核反应生成OOH,中间伴随电子转移(M–O+OH⁻→M–OOH+e⁻)。
最后,OOH被OH⁻去质子形成O₂并释出,表面位点重新生成OH完成循环(M-OOH+OH⁻→M–OH+O₂↑+e⁻)。可以看出,碱性介质下每步反应消耗OH⁻且释放电子,同时产生水而无自由质子累积。
酸性与碱性路径在吸附中间体种类上都是 OH、O、OOH,但质子在体系中的角色截然不同:酸性下质子作为产物被不断移走,碱性下则通过OH⁻提供质子并生成水。此外,电催化表面在碱性条件往往天然被–OH覆盖,初始状态常是M–OH(而非裸金属),因此有时将第一步视为 *OH的进一步脱氢过程。
总的来说,两种介质的基元步骤在本质上类似,但酸性OER通过质子释放平衡电荷,而碱性OER通过OH⁻供给质子并生成水来实现电荷平衡。
AEM与LOM机制的差异:值得注意的是,在不同催化剂和条件下,OER可能遵循上述传统的AEM机制,亦可能涉及晶格氧参与机制(LOM)。AEM机制下氧来自于吸附的水/羟基,步骤都伴随质子-电子协同转移,而LOM机制中,氧的一部分来自催化剂晶格本身。
LOM一般发生在某些过渡金属氧化物/氢氧化物催化剂中,表现为高价金属可以抽取自身晶格氧参与O–O键形成。在LOM路径中,通常催化剂表面的–OH基团经多步失去质子转化为高价的=O或“O⁻”形式(即“晶格氧”被活化),然后两个相邻晶格氧(或一个晶格氧与一个吸附氧物种)直接发生O–O耦合生成O₂,这一步不需要额外电子转移,属于非电化学的化学脱附过程。
随后催化剂晶格中留下的氧空位会被溶液中的OH⁻重新填补,以完成催化循环。这种LOM机制打破了每步都需质子-电子同步转移的限制,被认为可绕过某些AEM路径中能垒较高的步骤,从而提高活性。
然而,LOM往往要求催化剂能够形成并稳定高价态(金属–氧键较弱、氧空位易形成)。酸性环境一般更倾向于AEM机制,因为大多数催化剂在酸中不宜形成氧空位且晶格氧一旦流失易引发结构溶解。
但在碱性环境下(尤其是NiFe-LDH等金属氢氧化物催化剂中),研究发现LOM机制可能占重要地位。例如,NiFe-LDH在高氧化电位下能够发生晶格氧的氧化和再生,即通过LOM路径提升OER活性。
OER自由能台阶图的构建方法
自由能台阶图的构建:自由能台阶图(又称Gibbs自由能梯阶图)是分析OER机理和活性的重要理论工具。其构建基于密度泛函理论(DFT)计算得到的各中间体吸附自由能。
典型做法是采用诺斯科夫等提出的“计算氢电极”(Computational Hydrogen Electrode, CHE)方法,以标准氢电极平衡条件作为参考来计算各步反应的自由能变ΔG。具体而言,在理论上将每一步的质子+电子转移等效为½H₂(g)的生成/消耗,从而避免直接处理显式的带电体系。
计算步骤包括:首先,在模型催化剂表面的活性位上优化吸附各种中间体(OH, O, OOH)的构型并计算其吸附能;然后引入零点能和熵校正得到自由能;最后,将这些自由能值按照反应进程排列绘制成台阶状的能量图。
横轴表示反应坐标依次经历4个基元步骤,纵轴为每一步后的相对自由能。通常在标准平衡电位1.23V(RHE)下考察各步ΔG:理想催化剂在1.23V应使4个步骤的ΔG都为零,从而无需过电位即可驱动反应。
但实际催化剂由于中间体键强的限制,往往无法同时满足OH、O、OOH的最佳吸附能,必然存在某一步ΔG为正值,即为潜在决定步骤(Potential-Determining Step, PDS)。
该步对应的自由能壁垒即表示需要施加的理论过电位η (η=最大ΔG/e–1.23V)。举例来说,许多过渡金属氧化物催化剂存在OH和OOH吸附能的线性关联,导致两者能量差约为3.2eV。
这使得至少有一步ΔG≈3.2eV/每O_2,即需要约0.37V的最低过电位,这被认为是传统AEM路径下OER的活性上限。如果催化剂能打破这种限制(例如通过LOM路径),则有望降低最大自由能垫高,从而降低过电位。
判断速率控制步骤和活性:自由能台阶图直观揭示了各步反应的热力学难易程度。速率控制步骤(RDS)通常对应图中自由能攀升最高的那个步骤。在1.23V条件下,若某一步ΔG远高于其它步骤,则该步骤反应最难发生,因而限制总体反应速率。
例如,在许多催化剂上,OH脱氢为O是最困难的一步,因此被视为RDS,实验上也常通过分析塔菲尔斜率或反应级数验证这一判断(见下节塔菲尔分析)。
另一方面,自由能图还能用于比较不同催化剂的理论活性:最大ΔG越小,理论所需过电位越低,催化剂活性越高。研究者常通过调整催化剂组成和结构来改变中间体的ΔG,从而优化自由能分布使各步ΔG更加平衡。
这种思路在理论上对应火山曲线的概念:过弱或过强的中间体吸附都会导致某步ΔG过大,只有达到适中吸附强度才能接近理想自由能分布。因此,自由能台阶图不仅帮助确定哪一步是限速步骤,也提供了改进催化剂的方向——即针对限速步骤调整相关中间体的键合强度。
需要强调的是,自由能分析反映的是反应热力学驱动力,假设各步均无较高的动力学屏障。实际反应中还需考虑各步的活化能,但一般自由能最高的步骤往往也对应最高的动力学障碍,因此自由能图对RDS的判断在大多数情况下是合理的。
DOI:10.1002/adma.202209307
典型OER数据图的构成与解读
理论研究OER机理和催化活性,常结合多种数据图表以从不同角度揭示关键信息。以下列举典型图像类型及其含义:
ΔG中间体自由能图:如上节所述,这是将4个OER基元步骤的吉布斯自由能变化绘制而成的柱状或折线图。每个台阶高度代表在某一给定电位下,从前一中间体转变为下一中间体的自由能变ΔG。
通过该图可以直接看出哪个步骤ΔG最大(限速步骤)以及该催化剂在该电位下的理论过电位是多少。
DOI: 10.1126/science.adk9849
电子态密度(DOS)与投影态密度(PDOS)图:DOS描述了材料中电子能级的分布情况,即每个能量下存在多少电子态;PDOS则将总态密度投影到特定的原子或轨道,以解析不同元素或轨道对电子态的贡献。
对于电催化剂而言,DOS/PDOS图是理解电子结构-催化性能关系的关键工具。例如,过渡金属氧化物催化剂的金属d带与氧2p带的位置关系被认为决定了M–O键强度,从而影响OER中间体的吸附能。
在PDOS图中,研究者常关注以下特征:(i)d带中心:过渡金属d轨道态密度的重心位置。d带中心较高(接近费米能级)通常意味着金属与氧吸附物的键合较强,而d带中心较低则键合较弱。
优化的d带中心可使中间体吸附强度适中,从而提高OER活性。(ii)O 2p带中心:氧2p态密度重心相对于费米能级的位置。
特别在晶格氧机制中,O2p能带位置被用作氧活性的判据:O2p能级越接近费米能级,意味着氧的价带电子更容易被抽出,晶格氧越容易被氧化参与O₂生成。例如,有研究指出,O2p带中心上移(更接近费米能级)会使晶格氧易于失去电子形成氧空位,即促进LOM发生。
(iii)反键态占据情况:通过PDOS可以观察金属–氧键的反键轨道是否在费米能级下被占据。反键态占据多表明金属–氧键中的电子已填充到不利于键合的轨道,暗示键强下降、更易断裂。
例如,NiFe-LDH中掺入某些杂原子(如Mo)后,发现Ni3d与O2p轨道杂化增强,在费米能级附近的反键态占据增多,导致Ni–O键变得更弱,更易形成氧空位,实现晶格氧参与。
总之,通过DOS/PDOS分析,可以定性了解电子结构的变化如何影响中间体吸附及反应路径。
例如,当发现掺杂使金属d与氧p的态密度重叠增加、反键态填充提高时,往往预示晶格氧活性提高,有利于LOM;而若d带中心明显下移,则可能导致吸附物过弱,活性下降。这些电子结构指标为理性设计催化剂提供了指导。
差分电荷密度图(Charge Density Difference, CDD):这是用来可视化反应过程中电子重新分布的图像。差分电荷密度通常定义为体系在发生相互作用后的电荷密度减去各组分(如吸附前的催化剂表面和气相中间体)的电荷密度之和,即Δρ=ρ(催化剂+吸附物)– ρ(催化剂裸表面)– ρ(吸附物气相)。
通过绘制Δρ的等值面或截面图,可以直观地看到电子在哪些区域发生了累积或缺失。图中常用两种颜色(例如黄色表示电子积累,青色表示电子亏缺)来标识相对于非相互作用状态的电荷变化。
解读方法:差分电荷密度图可以告诉我们键形成或断裂时的电荷转移方向和程度。例如,在OH吸附在金属位时,差分电荷密度可能显示氧原子周围出现电子积累(来自金属提供的价电子),而金属原子与氧之间区域呈电子亏缺,表示金属向氧提供电子形成共价键。
结合定量的Bader电荷分析,可以进一步获得电子转移的数值大小,但差分电荷密度图本身已足以提供反应过程中电子行为的图像化理解。
DFT理论计算获取OER
计算方法与软件:理论上获取上述自由能和电子结构信息,主要依赖于密度泛函理论(DFT)计算。常用的软件包括VASP、QuantumESPRESSO、Gaussian(配合嵌入模型)、CP2K等。
其中VASP因擅长周期性体系的平面波模拟,被广泛用于固体催化剂表面吸附和反应能垒计算。一般流程是:首先构建催化剂的表面模型,如NiFe-LDH常用其最稳定的晶面构建有限层数的剖面(比如(001)晶面)并在两侧加真空层以模拟表/界面。
然后选择合适的交换相关泛函(常用泛函如PBE),针对过渡金属含量高的体系往往还需引入DFT+U方法校正d电子的自相互作用(例如Ni和Fe的3d轨道通常施加一个U参数以更准确描述Ni^3+/Fe^3+高自旋态)。
接着在所选表面活性位上吸附中间体(OH, O, OOH等),对吸附构型进行几何优化以找到最低能量构型。同时考虑自旋极化计算,因为Ni、Fe等过渡金属可能具有未成对电子,强磁性态会影响吸附能和电子结构。
对于NiFe-LDH这种含Fe^3+的体系,初始可以给Fe设定高自旋态,以模拟实验观察到的Fe在NiOOH中的低自旋/高自旋转变行为。
模型构建要点:构建合理的计算模型对可信结果至关重要。以NiFe-LDH为例,它在静态条件下为层状双氢氧化物,中间插有阴离子(如NO₃⁻或CO₃²⁻)平衡电荷。然而在强氧化电位下,NiFe-LDH实际转变为Ni、Fe氧羟化物活性相(类似于NiOOH/FeOOH)。
因此理论计算时通常直接采用NiFeOOH晶格模型:比如以β-NiOOH的层状结构为基底,在Ni位替换一定比例的Fe,从而构造Ni:Fe约3:1(25%Fe)的模型来代表NiFe-LDH的活性相。这样的模型捕捉了Fe掺杂对Ni氧化物的电子结构影响,同时简化了插层阴离子的影响(假定在高电位下阴离子已脱出或作用可忽略)。
模型中需确保整体电荷中性(通常每引入一个Fe^3+取代Ni^2+,需在层间加一额外OH⁻或调整总体H含量)。此外,模型尺寸要足够大以避免吸附物之间的镜像干扰;通常采用2×2或更大的超胞,并在垂直方向加>15Å真空。
溶剂效应也是一大考虑因素:纯DFT为零温真空中的能量,需要对水溶液环境下自由能进行校正,例如加上中间体的溶剂化能近似或采用连续介质模型(如VASP-Sol等)模拟水的介电屏蔽。
自由能计算与过电位评估:在获得吸附态的DFT总能量后,还需考虑零点振动能(ZPE)和熵(S)修正以得到室温下的Gibbs自由能:G = E_DFT + ZPE – T·S。其中ZPE和S可通过计算吸附物和气相物种的振动频率来估算(通常小分子可以直接计算频率,大体系可近似各局部振动模式)。
特别地,气相H₂的G(T)可由热力学数据获得。在酸性条件下,质子-电子对以½H₂参考,则在标准条件(298K,pH0)下½H₂的G = 0,因此各步ΔG = ΔE_DFT + ΔZPE – TΔS – eU(其中eU项表示施加电位U对每转移1e⁻的自由能降低)。
通过在U=1.23V下计算ΔG,可以判断是否有步骤仍为正。如果有,说明该催化剂需要额外过电位η使该步变为平衡,否则此正的ΔG即为导致过电位的热力学瓶颈。这样的分析重复于不同材料,可预测其相对活性高低。
DOI:10.1002/smll.202304260
总结
通过以上分析,我们了解到酸性和碱性条件下OER机理的差异源于质子参与方式和催化剂电子结构的不同。酸性介质中反应遵循质子-电子协同的吸附质演化路径,而碱性介质中OH⁻的参与引出更复杂的机制,尤其是在NiFe-LDH等催化剂中可能出现晶格氧直接参与产氧的LOM路径。
自由能台阶图是理论研究OER的重要工具,它量化了各步反应的热力学要求,可用于判定限速步骤和比较不同催化剂的活性。
事实证明,深入的理论计算与图像分析在电催化研究中已成为不可或缺的一环,它为理解反应本质和催化剂改性提供了清晰的“显微镜”,也为我们走向理性能源催化设计时代奠定了基础。

点击阅读原文,立即下单!?