本文华算科技系统介绍了自旋密度的基本概念及其在凝聚态物理和材料科学中的重要性。自旋密度是描述材料中电子自旋分布的关键物理量,通过分析自旋向上和自旋向下电子的密度差异,可以揭示材料的磁性特征和电子行为。
文章详细阐述了自旋密度的定义、计算方法及其在铁磁性、反铁磁性和亚铁磁性材料研究中的应用。自旋密度的空间分布直接影响材料的磁学、电学和光学性质,为理解材料的宏观性能提供了微观理论基础。
此外,本文还探讨了自旋密度在自旋电子学领域的潜在应用价值,为相关研究提供了理论支持。
自旋密度基本概率
在量子力学中,电子不仅具有电荷属性,还具有内禀角动量,即自旋。自旋是电子的固有属性,其取值为±½,通常用向上(↑)和向下(↓)的箭头表示。自旋密度则是描述材料中电子自旋分布的物理量,它反映了不同自旋方向电子在空间中的密度分布情况。

对于一个多电子体系,设α自旋态(自旋向上)的电子波函数为ψα(r),β自旋态(自旋向下)的电子波函数为ψβ(r),则自旋密度ρs(r)的定义为:ρs(r)=ρα(r)−ρβ(r)。
其中,ρα(r)= | ψα(r)|2是α自旋态电子的密度,ρβ(r)= | ψβ(r)|2是β自旋态电子的密度。这个定义式清晰地展现了自旋密度的物理本质:它表征了空间某点处两种自旋态电子分布的不平衡程度。
在实空间分析中,自旋密度的数值特征具有明确的物理意义:当某点的自旋密度值为正时,表明该空间位置自旋向上的电子密度占据优势;反之,若为负值,则说明自旋向下的电子密度更大。
特别值得注意的是,对于闭壳层电子体系,由于每个α电子都有对应的β电子与之配对,二者密度在空间各点完全相等,因此体系的自旋密度处处为零;而在开壳层体系中,由于存在未配对电子,自旋密度呈现非零分布,通过三维自旋密度图可以直观展示未配对电子在空间中的具体分布特征。
从积分性质来看,对自旋密度函数在全空间范围内进行积分,其结果等于体系内α电子数与β电子数的差值,这个差值定量反映了整个体系的自旋极化程度。
自旋密度在凝聚态物理和材料科学领域具有重要的应用价值,特别是在材料的磁性研究方面起着决定性作用。从微观机制来看,材料的磁性本质上源于电子的自旋磁矩和轨道磁矩的共同贡献,其中自旋密度的空间分布模式直接决定了材料中磁矩的大小、方向和有序性。
具体而言:在铁磁性材料中,自旋密度分布表现出长程有序的特征,某些区域的电子自旋呈现一致排列,导致材料表现出宏观自发磁化现象;在反铁磁性材料中,相邻原子格点的自旋密度分布呈现反平行排列,使得净磁矩相互抵消;而在亚铁磁性材料中,自旋密度的反平行排列存在不完全抵消,表现出特殊的磁学性质。
此外,自旋密度还与材料的诸多物理性质密切相关:在电学性质方面,自旋密度分布影响载流子的自旋极化输运;在光学性质方面,自旋密度差异会导致自旋选择的光学跃迁;在热学性质方面,自旋密度波动的特征与材料的磁热效应直接相关。
特别值得强调的是,在快速发展的自旋电子学领域,对自旋密度的精确描述和调控具有关键性的技术意义。研究人员通过第一性原理计算和先进表征手段获取自旋密度的详细分布信息,为设计新型自旋电子器件(如自旋阀、磁隧道结、自旋场效应晶体管等)提供了理论基础。
同时,对自旋密度与材料性能关联规律的深入理解,也为开发高性能磁性存储材料、量子计算元件等前沿应用指明了方向。因此,自旋密度研究不仅具有重要的理论价值,更在新型功能材料的设计和开发中展现出广阔的应用前景。
案例分析1
图中DFT计算自旋密度在研究氧析出反应(OER)催化剂NiₓFe₁₋ₓOOH的机理中具有核心意义,它从量子力学的角度揭示了自旋相关的电子转移过程如何影响催化活性,并为理解过渡金属基催化剂的构效关系提供了关键证据。
通过分析自旋密度分布、轨道相互作用和电子结构特征,这项工作不仅阐明了Ni-Fe协同作用的本质,还为设计高效自旋电子催化剂提供了理论框架。
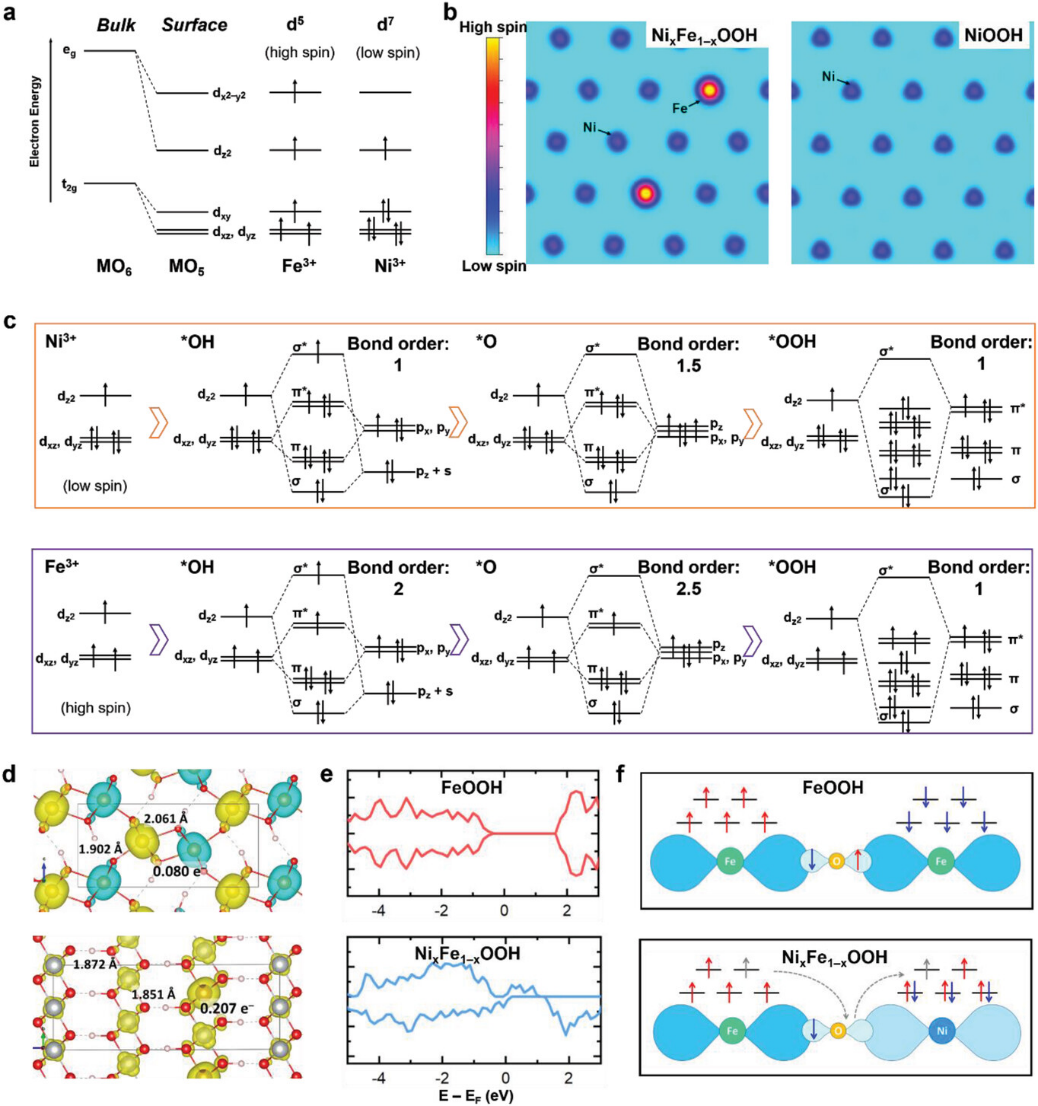
自旋密度的空间分布直观反映了催化剂中未配对电子的局域化特征,这对于理解电荷传输和反应位点的活性至关重要。在图d中,纯FeOOH的自旋密度图显示其反铁磁(AFM)耦合导致相邻Fe³⁺离子的自旋方向相反,使得连接氧原子(Fe–O–Fe)的自旋密度显著降低(仅0.080 e⁻)。
这种自旋抵消效应会阻碍电子在材料中的长程迁移,导致FeOOH导电性差,这与实验中观察到的OER活性较低现象一致。相比之下,NiₓFe₁₋ₓOOH(x > 0.7)中铁磁(FM)耦合的高自旋Fe³⁺与低自旋Ni³⁺通过氧桥(Fe–O–Ni)形成自旋极化通道,其自旋密度高达0.207 e⁻。
这种自旋非对称性使得电子能够沿特定方向高效传输,从而显著提升催化剂的整体导电性。图e的态密度(DOS)分析进一步支持这一结论:FeOOH因AFM耦合存在明显的带隙,而NiₓFe₁₋ₓOOH呈现半金属特性,其自旋向上通道跨越费米能级,为电子转移提供了无阻碍路径。
自旋密度的差异还直接关联到催化反应的热力学与动力学行为。图c通过键级分析揭示了Fe³⁺和Ni³⁺位点与反应中间体(OH、O、OOH)的轨道相互作用差异。高自旋Fe³⁺的d电子构型(t2g³eg²)使其eg轨道与OH的σ轨道形成更强的共价键(键级为2),而低自旋Ni³⁺(t2g⁶eg¹)的键级仅为1。
这种差异导致Fe³⁺位点对OH的吸附更强,更易于启动OER循环;同时,Fe³⁺对OOH的吸附较弱(键级降低),有利于产物O₂的释放。这种“强吸附–弱脱附”的平衡符合Sabatier原理,解释了为何Fe³⁺是OER的活性中心。
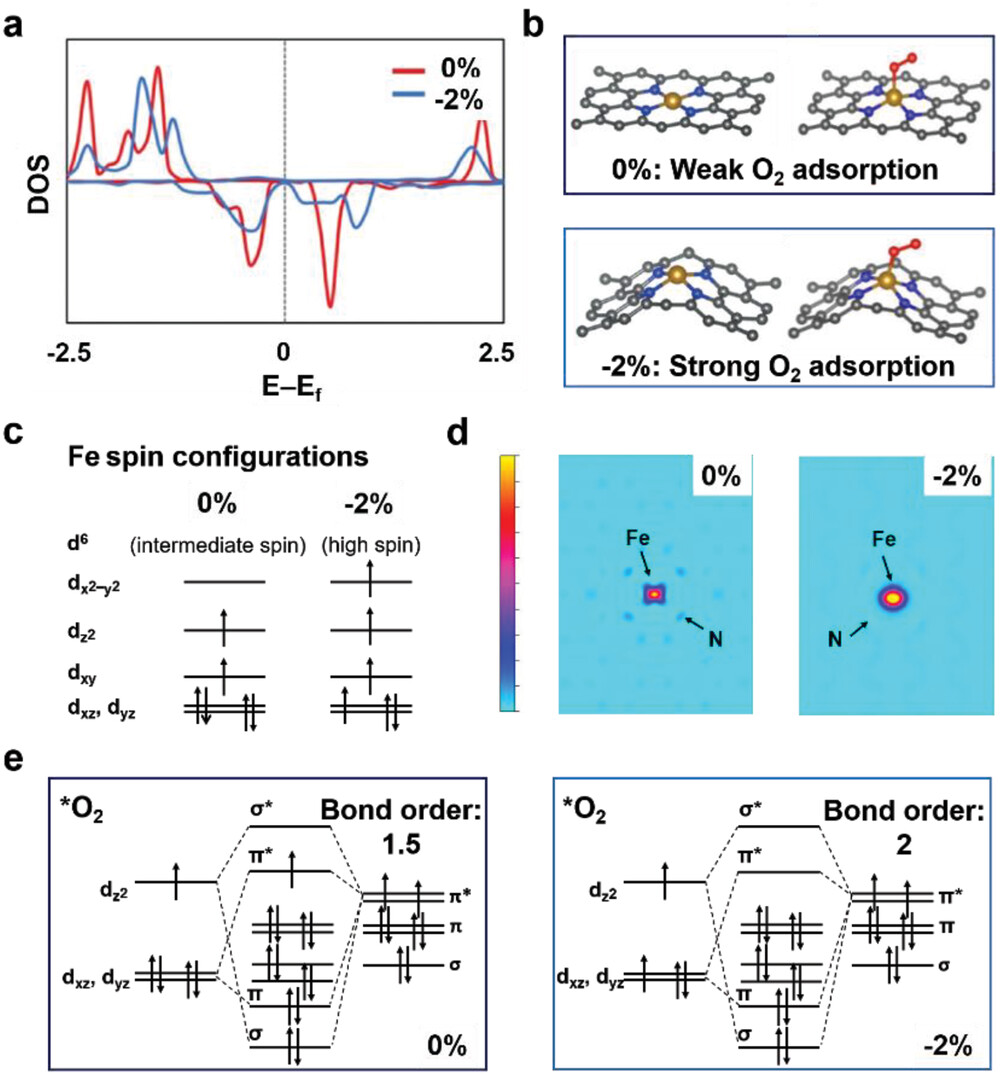
此外,自旋相关的电子转移还通过量子自旋交换相互作用(QSEI)调节反应能垒。例如,在ZnCo₂O₄和ZnMn₂O₄中,不同自旋态会改变速率决定步骤(RDS),进一步证明自旋构型对反应动力学的调控作用。
研究的创新性在于将自旋自由度引入传统电催化理论框架,挑战了仅从热力学(如吸附能)或几何结构(如配位环境)解释活性的局限。例如,此前关于NiₓFe₁₋ₓOOH活性位点的争议中,实验曾提出Fe³⁺作为Lewis酸促进Ni⁴⁺生成,或Ni-OO⁻物种主导反应等观点,但均未涉及自旋效应。
而本工作通过自旋密度和DOS分析表明,Fe³⁺的高自旋态不仅优化了电子传输,还通过自旋选择性电子转移降低了氧分子(三重态O₂)生成的势垒。这一机制在实验中得到佐证:外加磁场可通过取向磁矩增强OER活性,且FM耦合的氧化物通常比AFM材料更具催化优势。
此外,自旋密度分析还为催化剂设计提供了明确方向。例如,通过调控过渡金属的价态和配体场可改变其自旋态(如Co³⁺在LaCoO₃中随粒径从低自旋转变为高自旋),从而优化eg轨道填充数;在单原子催化剂FeN₄中,Fe-N键收缩可诱导高自旋态增强O₂吸附能力。
这些案例均表明,自旋工程是超越传统电子结构调控的新策略。未来,结合原位自旋表征技术(如自旋极化STM或X射线磁圆二色性)和多尺度模拟,有望进一步揭示动态反应条件下自旋序的演化规律。
总之,自旋密度分析不仅深化了对Ni-Fe协同催化机制的理解,还确立了自旋相关电子转移在氧电催化中的普适性角色。
这项工作为开发基于自旋调控的高效催化剂奠定了理论基础,并推动电催化研究从“电子结构”范式向“自旋–轨道–电荷”多自由度耦合范式拓展。通过精确设计材料的自旋序(如FM耦合、高自旋态)和界面自旋极化,未来或可实现超越Pt/Ir基催化剂性能的廉价材料体系,助力清洁能源技术的发展。
案例分析2
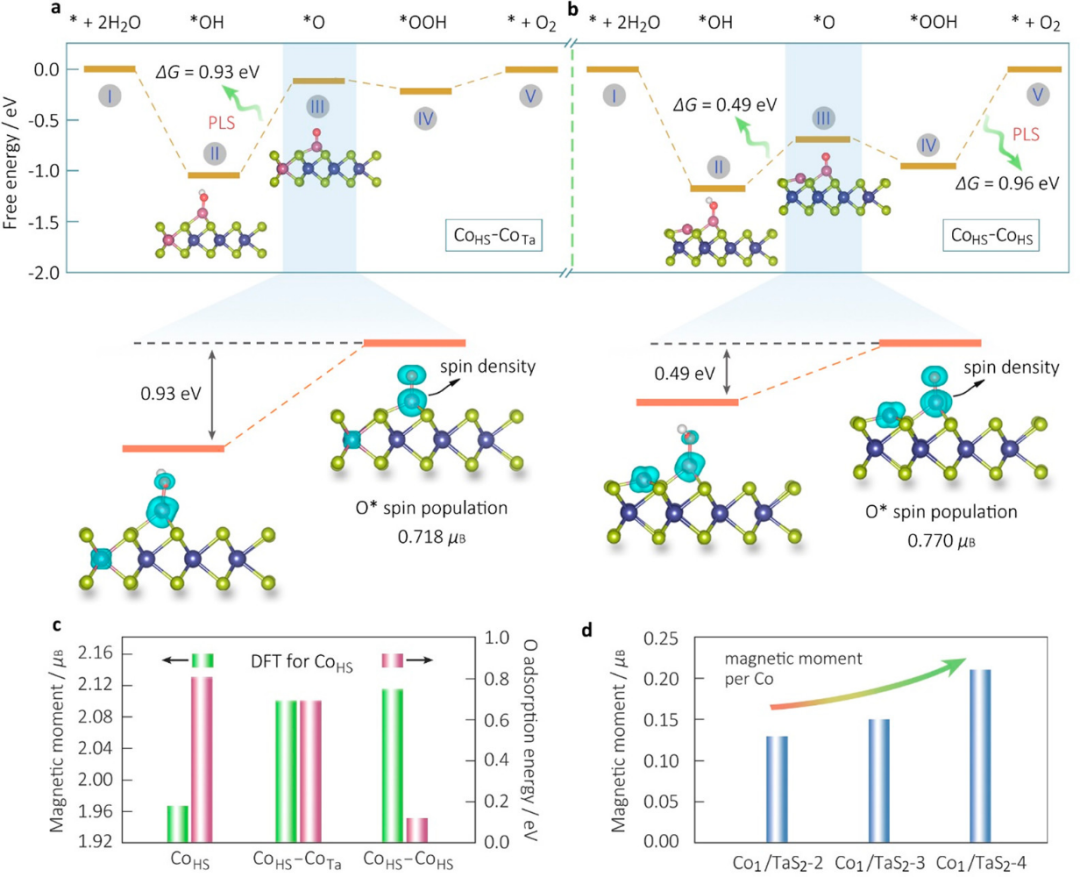
在下图中,通过自旋密度分析深入揭示了钴单原子催化剂(Co₁/TaS₂)中自旋态与析氧反应(OER)活性之间的关键关联,为理解磁性单原子催化剂的活性起源提供了重要理论依据。
研究首先通过计算发现,CoHS位点的自旋密度可以通过相邻CoHS或CoTa位点的交换相互作用进行调控,而这种调控直接影响了氧中间体(*O)的结合能,进而决定了OER的活性。
具体而言,当CoHS位点与CoTa位点相邻形成CoHS–CoTa双位点时,其自旋密度被适度增强,导致氧中间体的结合能达到最优值,从而显著降低了反应能垒(0.93 eV),提升了OER活性。
这一发现表明,适中的自旋密度能够有效优化催化过程中的关键步骤(如*OH → O的转化),为设计高效催化剂提供了新的思路。
然而,当CoHS位点与另一个CoHS位点相邻形成CoHS–CoHS双位点时,自旋密度过度增加,导致CoHS-O结合过强,反而阻碍了后续反应步骤(如*OOH的脱附),使得反应能垒升高至0.96 eV,抑制了OER性能。这一结果清晰地展示了自旋密度与催化活性之间的非线性关系,强调了自旋态调控在催化反应中的重要性。
进一步分析发现,这种自旋密度的调控机制主要依赖于磁性原子之间的RKKY相互作用。相邻的CoTa位点通过长程自旋交换作用增强了CoHS位点的自旋极化,形成稳定的O自由基,从而促进了OER的关键步骤。
相比之下,相邻的CoHS位点则因过度自旋极化使得氧中间体过于稳定,导致反应动力学受阻。这种机制不仅解释了实验中观察到的OER活性变化趋势,还为磁性催化剂的理性设计提供了理论指导。
实验数据与理论计算的高度一致性进一步验证了这一机制的可靠性。例如,磁化测量结果显示,随着Co负载量的增加,单个Co原子的平均磁矩从0.13 μB(Co₁/TaS₂-2)逐渐增加到0.21 μB(Co₁/TaS₂-4),与DFT预测的自旋密度变化趋势完全吻合。这种理论与实验的协同验证不仅强化了自旋密度作为活性描述符的可信度,还为后续研究提供了坚实的科学基础。