

说明:态密度(DOS)是凝聚态物理和材料科学中的核心概念,描述单位能量区间内可用电子态的密度,是连接微观电子结构与宏观物性(如导电性、光学吸收、催化性能)的关键桥梁。
不同维度材料(1D、2D、3D)因色散关系不同,其态密度表现出明显差异。文中深入探讨了态密度在电催化领域的应用,尤其在活性位点识别、电荷转移机制、缺陷与掺杂效应、以及机器学习高通量筛选中的价值。
计算方面,以密度泛函理论(DFT)为核心,通过选择不同泛函、赝势、k点等参数精确构建材料DOS。通过Ga₆N₆纳米环的案例,文中华算科技展示了DOS分析在气体吸附与传感中的关键作用。态密度分析已成为材料设计与性能优化不可或缺的理论工具。
什么是态密度?
态密度(DOS)作为凝聚态物理和材料科学中的核心概念,用于描述单位能量区间内单位体积的量子态数量,其数学表达式为DOS (E)=dN/dE,其中N为能量小于E的量子态总数。
这一物理量通过揭示材料中电子能级的分布规律,直接关联电荷填充、电导率、光学响应等宏观性质,是连接微观电子结构与宏观材料性能的关键桥梁。
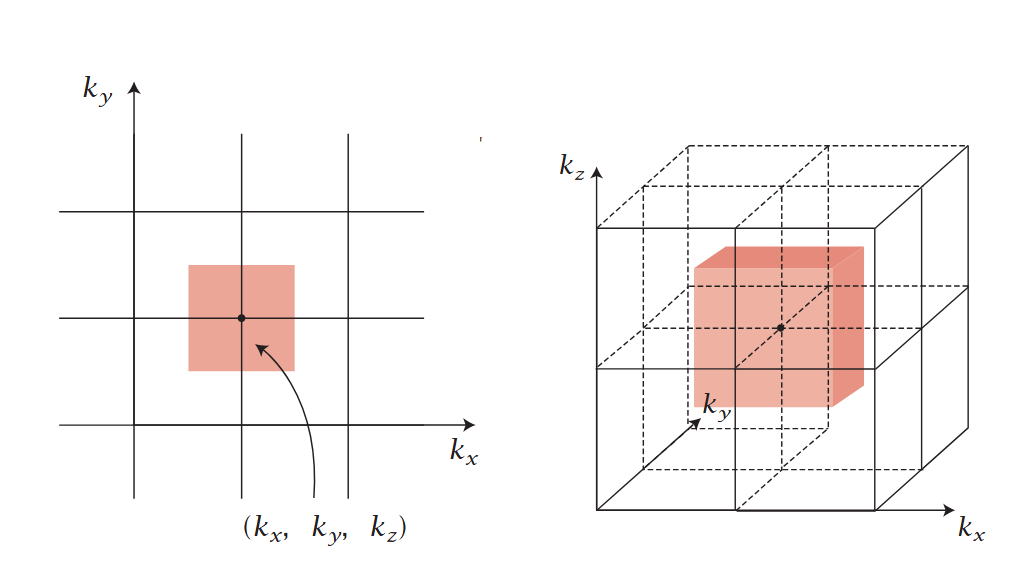

DOI:hdl.handle.net/2345/2886
从数学定义来看,在连续能带体系中,态密度通过动量空间的积分获得,即DOS (E)=(1/(2π)³)∫δ(E – ε(k)) dk,其中ε(k)为色散关系,描述能量与波矢k的关系;对于无序系统(如非晶材料),则需在热力学极限下通过测度收敛求解,以避免因结构无序导致的态密度波动。
态密度的特性具有显著的维度依赖性:三维材料中,自由电子气模型呈现抛物线色散关系(ε(k) ∝ k²),对应的态密度随能量平方根增长,如金属铜的导带态密度在费米能级附近呈现平滑分布;二维材料(如石墨烯)因线性色散关系(ε(k) ∝ |k|),态密度在狄拉克点附近呈现V形特征(DOS (E) ∝ |E|),这种线性态密度使石墨烯具有高载流子迁移率;一维体系(如碳纳米管)的态密度则表现为DOS (E) ∝ 1/√(E – E₀),在范霍夫奇点处出现发散,这种强烈的能态集中使其在特定能量下表现出显著的光学吸收特性。
DOI:10.1007/s40766-023-00043-8
态密度的物理意义体现在多个方面:费米能级(E_F)处的态密度直接决定电子热容和电导率,例如贵金属Pt因DOS较高,具有优异的导电性能;低维材料在带边的高态密度可增强光吸收和载流子分离效率,如二维MoS₂的价带顶态密度是体相材料的3倍,使其光催化活性显著提升。
此外,态密度的分布特征(如峰值位置、宽度)还能反映电子的局域化程度,宽而平的态密度表明电子离域性强,窄而尖的峰值则对应电子局域化。
综上,态密度通过量化电子能态的分布特征,为理解材料的电学、光学和催化性能提供了不可或缺的理论工具。
DOI:10.1016/j.jallcom.2025.182547
态密度分析应用
在电催化领域(如ORR、HER、CO₂RR)的顶刊研究中,态密度分析作为解析催化剂活性起源的核心手段,通过揭示电子能级分布与反应性能的关联,为催化剂设计提供了原子级指导,其应用主要聚焦于活性位点电子结构、电荷转移机制、缺陷与掺杂效应及高通量筛选四个方向。
活性位点电子结构解析中,d带中心理论是连接态密度与催化活性的重要桥梁,过渡金属催化剂的活性与d带中心位置(ε_d)密切相关,而ε_d通过态密度积分计算。
研究表明,d带中心向费米能级上移会增强反应物与催化剂的轨道杂化,提升吸附强度,但可能因中间体脱附困难降低反应速率,例如Pt₃Ni合金的d带中心比纯Pt低0.2 eV,削弱了OH的吸附,使ORR过电位降低0.15 V。
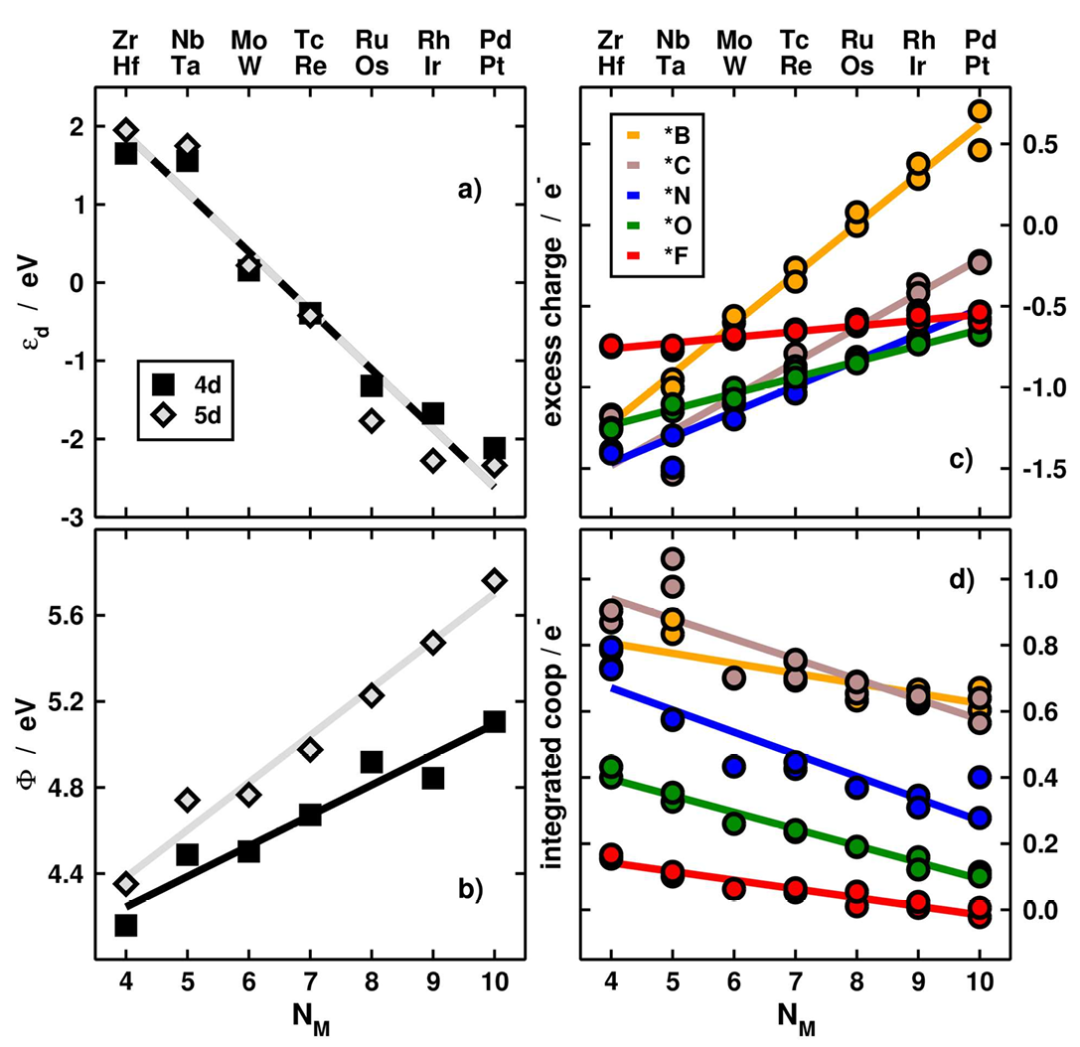
DOI:10.1021/acs.jpclett.6b02430
电荷转移机制的研究中,费米能级处的态密度反映催化剂的电荷注入能力,DOS越高,可参与反应的电子态越多,电荷转移阻力越小。
研究显示,Fe-N-C催化剂中FeN₄活性位点的DOS是无Fe位点的5倍,显著提升ORR动力学,半波电位达0.89 V vs. RHE;石墨烯基催化剂中,狄拉克点附近的线性态密度使O₂吸附能优化至-0.3 eV,避免过强或过弱吸附,CO₂RR的CO选择性达90%。
缺陷与掺杂效应通过态密度的变化得到直接证实,N掺杂碳材料的态密度在费米能级附近出现明显峰值,源于N原子引入的缺陷态电子,这些电子可与O₂的π轨道作用,增强ORR活性;Co₃O₄/石墨烯界面的态密度偏移显示,Co的3d轨道电子向石墨烯转移0.12 e⁻,这种电荷重分布使*OOH吸附能降低0.2 eV,加速OER进程。
态密度衍生参数作为机器学习模型的输入,实现了催化剂的高通量筛选,例如基于10⁴种过渡金属氧化物的态密度数据训练的模型,可预测OER过电位,误差,从候选材料中快速识别出CoFe₂O₄等高效催化剂。
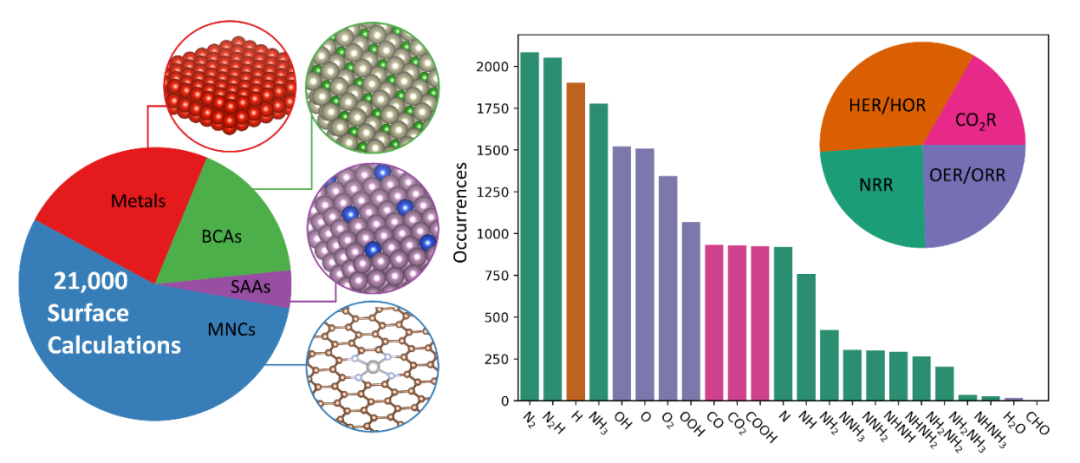
DOI:10.1021/acs.jpcc.4c06826
这些应用案例共同表明,态密度分析不仅能解释催化活性的起源,更能为催化剂的理性设计提供可量化的描述符,推动电催化研究从经验探索向理论驱动转变。
态密度怎么算?
态密度(DOS)的计算以密度泛函理论(DFT)为核心框架,通过合理选择计算参数、泛函与分析技术,实现对电子能级分布的精准量化,其流程与设置直接影响结果的可靠性与物理意义。
计算流程始于泛函类型的选择,不同泛函适用于不同体系:GGA-PBE泛函因平衡计算效率与精度,广泛用于金属、合金等催化体系的态密度计算,但其存在低估带隙的缺陷,如计算TiO₂的带隙时,PBE结果低于实验值。
GGA+U方法通过引入Hubbard U参数校正强关联体系(如过渡金属氧化物NiO、Co₃O₄)中d电子的局域化效应,例如对Co₃O₄进行设置,可使Co的3d态密度峰值更尖锐,准确反映电子局域性。
杂化泛函(如HSE06)通过混合25%的精确交换能,显著提高带隙计算精度,适用于半导体催化剂的态密度分析,但计算成本是PBE的5-10倍。
赝势方法的选择同样关键,投影缀加波(PAW)方法通过将电子分为芯区与价区,芯区用赝势描述,价区保留全电子特性,能精确处理价电子与芯电子的相互作用,在计算重元素的态密度时,PAW方法的精度显著高于ultrasoft赝势。
参数设置需兼顾收敛性与计算成本:k点网格用于布里渊区积分,二维材料推荐≥5×5×1,三维材料需≥8×8×8,以确保态密度的平滑性,避免因采样不足导致的伪振荡;截断能(Cutoff)设置为500–600 eV,防止平面波基组截断误差影响能级分布;Smearing宽度采用0.05–0.2 eV的Gaussian展宽,平衡态密度的分辨率与计算稳定性;自洽场(SCF)收敛标准需≤10⁻⁵ eV/atom,确保电子密度收敛,使态密度计算误差。
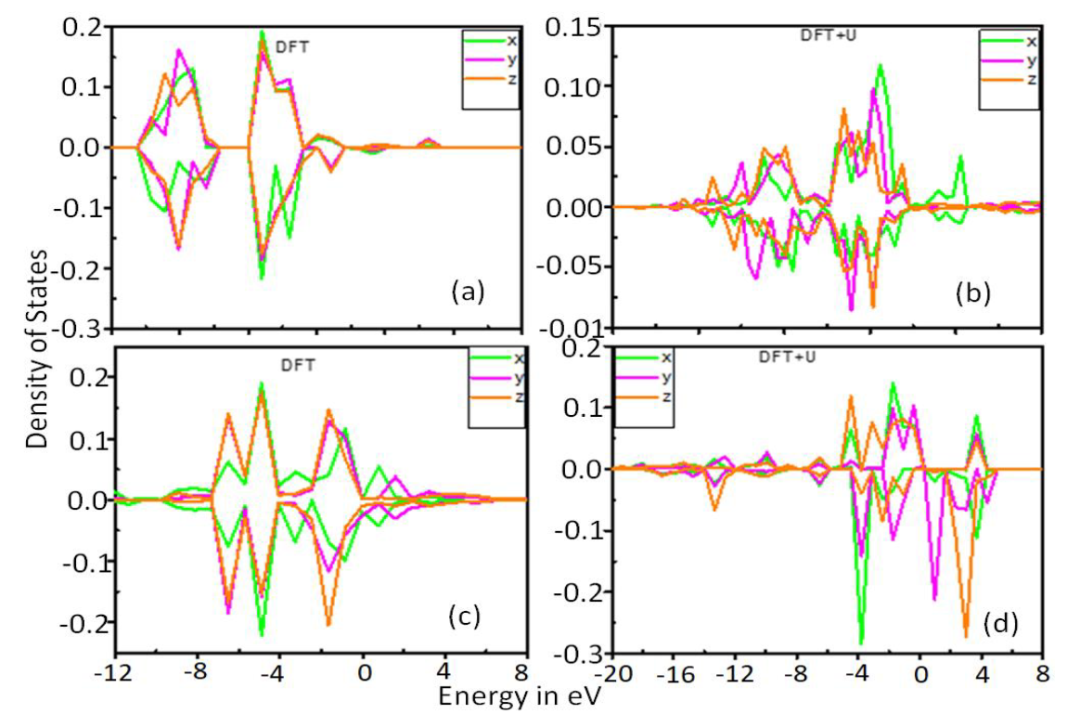
DOI:10.3329/bjphy.v27i1.49725
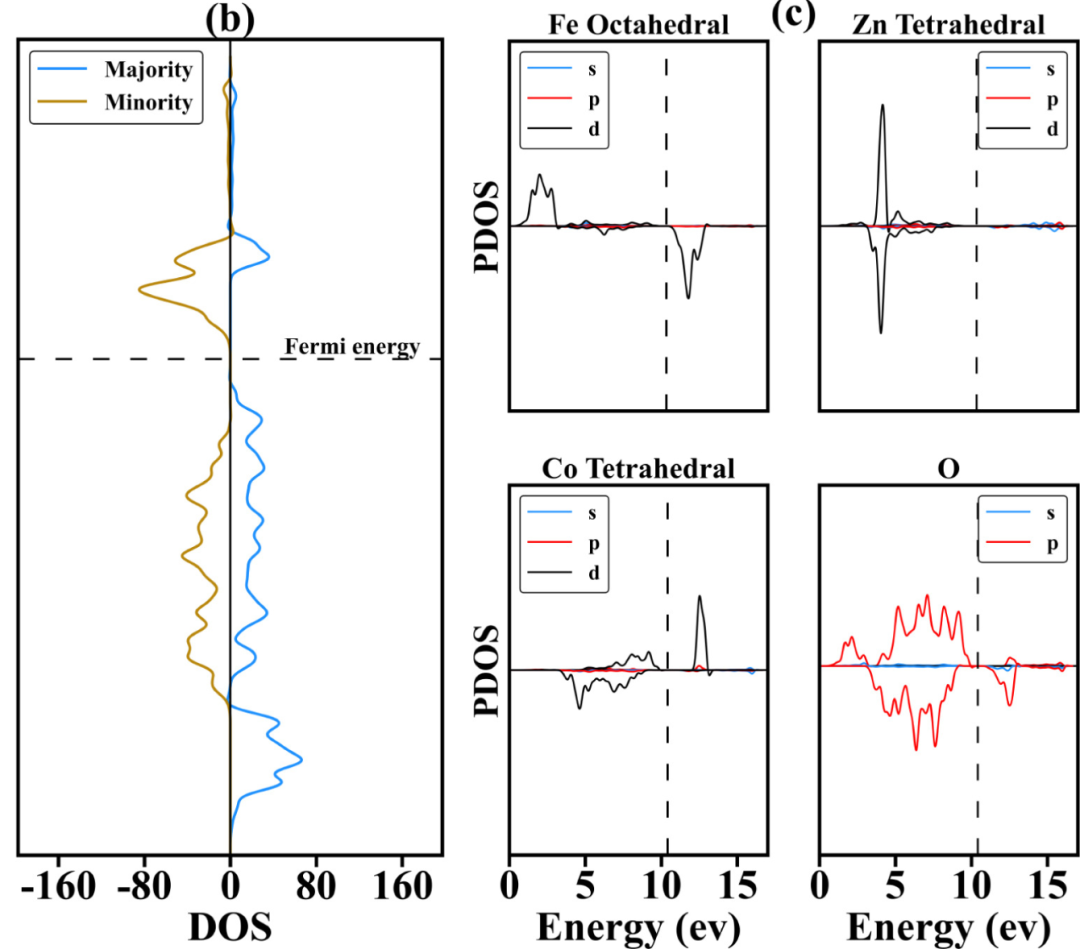
关键分析技术包括投影态密度(PDOS)与晶体轨道哈密顿布居(COHP):PDOS将总态密度分解为不同原子的s、p、d、f轨道贡献,可识别活性位点的关键轨道,如Fe-N-C催化剂的PDOS显示,Fe的3d轨道在费米能级附近有显著贡献,证实其为ORR活性中心;COHP通过分析键合态与反键态的占据数,量化轨道相互作用强度,例如COHP计算表明,Pt与*H的σ键合态占据率达80%,解释了适中的吸附能。
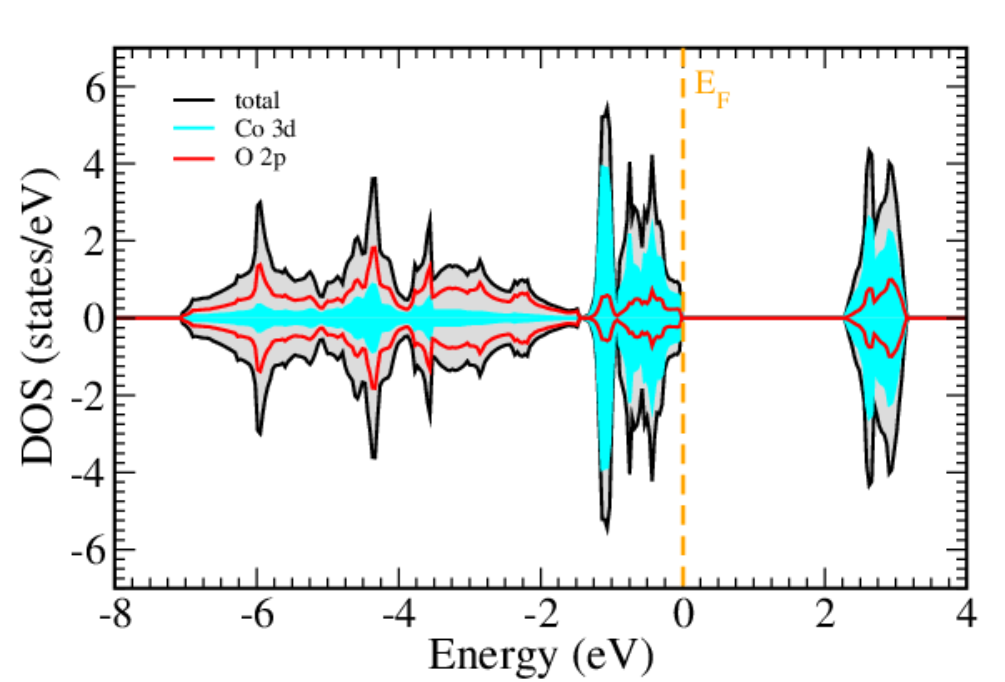
DOI:10.1103/PhysRevMaterials.6.040302
这些计算方法与分析技术的协同应用,使态密度从抽象的物理概念转化为可操作的量化工具,为理解催化机制与设计高效材料提供了坚实的数值支撑。
Ga₆N₆纳米环DOS研究
“Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst”使用了密度泛函理论(DFT)对Ga₆N₆纳米环的电子结构进行了深入研究,尤其是通过态密度(DOS)的分析,探讨了不同气体分子与该纳米环之间的相互作用,尤其是其吸附性能。
研究的核心是揭示Ga₆N₆纳米环的电子结构特征,特别是气体分子如何通过电子转移和轨道重叠与Ga₆N₆发生相互作用,从而改变其电学性质和吸附行为。
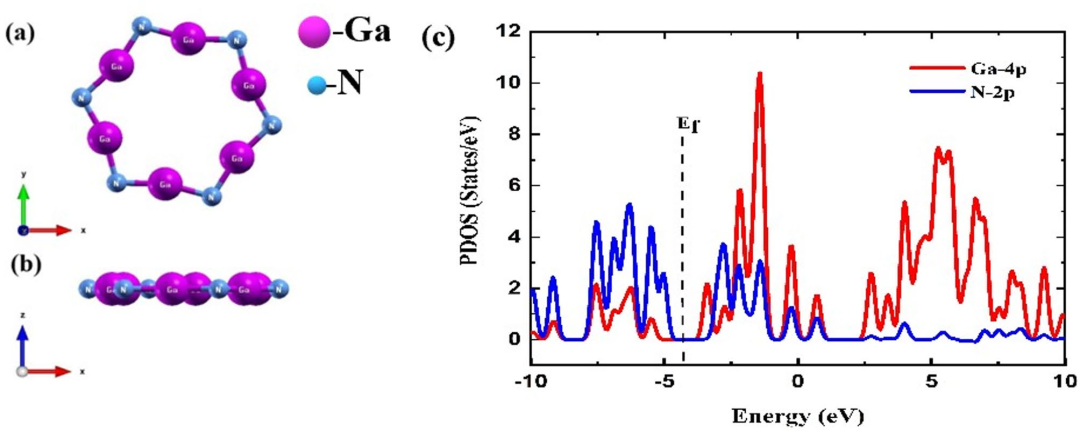
DOI:10.1038/s41598-025-06067-w
首先,研究团队对Ga₆N₆纳米环的几何结构进行了优化,得到了一些基本的结构参数,如Ga-N键长为2.01 Å,Ga-N-Ga的键角为105.35°,这些数据表明Ga₆N₆具有较高的稳定性。
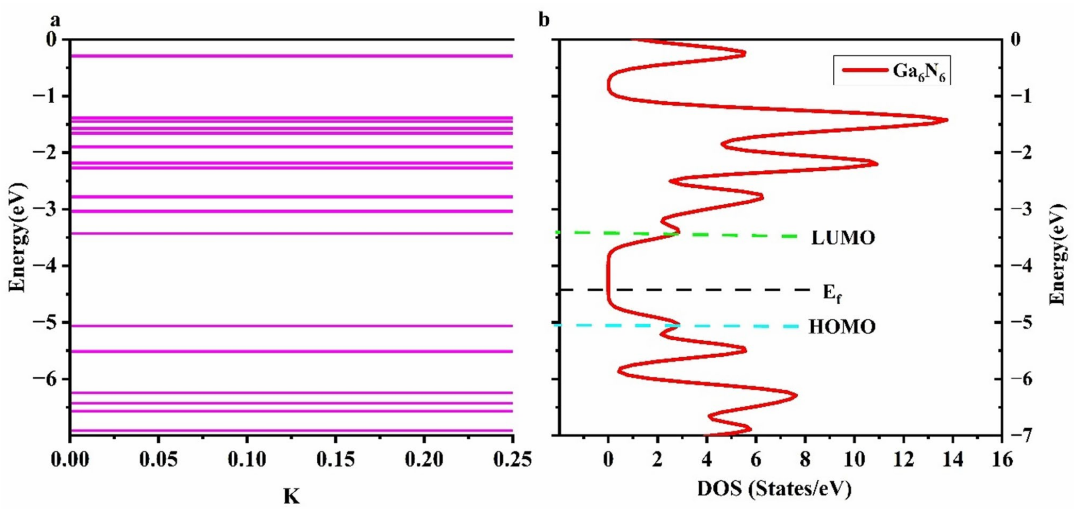
通过对该结构进行态密度分析,作者揭示了Ga₆N₆纳米环的电子结构。在该纳米环的最高占据分子轨道(HOMO)中,主要由氮原子的2p轨道贡献,而最低未占据分子轨道(LUMO)则主要由镓原子的4p轨道贡献。
根据这些分析,Ga₆N₆被归类为半导体,具有较小的能隙(约1.73 eV),这一特性使得Ga₆N₆在气体吸附过程中表现出较好的电子响应性。
在气体吸附方面,作者计算了多种气体分子(如NO、NO₂、SO₂、CO、NH₃等)与Ga₆N₆纳米环的吸附能。
研究结果显示,NO、NO₂和SO₂等气体表现出较强的化学吸附行为,吸附能分别为-1.75 eV、-2.04 eV和-1.01 eV,而CO和NH₃的吸附能则相对较低,表明它们主要通过物理吸附与Ga₆N₆表面相互作用。
这些结果表明,Ga₆N₆对某些气体分子具有较强的吸附能力,尤其是对于NO、NO₂和SO₂等有毒气体的捕获能力。
通过投影态密度(PDOS)分析,研究进一步揭示了气体分子对Ga₆N₆纳米环电子结构的影响。研究表明,NO和NO₂的吸附会导致Ga₆N₆的Fermi能级下移,表明吸附过程涉及较强的电子相互作用。
这一变化主要是由于气体分子和Ga₆N₆表面之间的轨道重叠,特别是在NO和NO₂吸附过程中,N原子的2p轨道与Ga原子的4p轨道之间的相互作用更为显著。这种电子结构的变化有助于增强气体分子与Ga₆N₆之间的吸附作用。
另外,文章还通过Hirshfeld电荷分析探讨了气体吸附过程中电荷转移的情况。NO、NO₂和SO₂等气体分子吸附后,Ga₆N₆纳米环表面发生了显著的电荷重分布,尤其是在NO和NO₂的吸附过程中,电荷转移现象更加明显。
这种电荷重分布通过产生有效的界面偶极子,进一步加强了吸附能量。这一过程表明,气体分子与Ga₆N₆纳米环之间的强电子相互作用是吸附过程的关键因素。
除了电荷转移,研究还通过分析前沿分子轨道(HOMO-LUMO)能级变化,进一步揭示了吸附气体对Ga₆N₆电子结构的影响。
特别是在NO₂吸附时,HOMO-LUMO能隙的缩小表明,吸附气体增强了Ga₆N₆的导电性,使得电子更容易从价带跃迁到导带。这种变化反映了气体分子与Ga₆N₆之间的强烈相互作用,进一步验证了吸附过程中的电子转移现象。
从吸附能的计算结果来看,Ga₆N₆纳米环对不同气体的吸附性能表现出了显著差异。
NO、NO₂和SO₂的较强吸附能表明该材料在捕获有毒气体方面具有较好的应用前景。尤其是在有毒气体的吸附过程中,Ga₆N₆不仅能够通过化学吸附有效捕捉气体分子,而且吸附过程还伴随着电子结构的显著变化,表明其具有较强的电子响应能力。
除了吸附能的计算,研究还探讨了气体分子吸附后Ga₆N₆的恢复时间(τ)。恢复时间是气体传感器性能的重要指标,决定了气体的吸附和脱附过程的速度。
研究发现,CO和NH₃的恢复时间较短,表明它们在Ga₆N₆表面的吸附是可逆的,而NO、NO₂和SO₂则显示出较长的恢复时间,这表明这些气体的吸附具有较强的持久性。这一结果为Ga₆N₆纳米环在气体传感和去除应用中的潜力提供了支持。
DOI:10.1038/s41598-025-06067-w
总的来说,Ga₆N₆纳米环在气体吸附、尤其是有毒气体的捕捉方面表现出了极大的应用潜力。通过态密度分析、电荷转移分析和吸附能的计算,研究揭示了Ga₆N₆与气体分子之间的相互作用机制,尤其是其电子结构在吸附过程中的变化。
未来,Ga₆N₆纳米环有望在环境保护、气体传感和气体净化等领域发挥重要作用。同时,研究还指出了进一步优化Ga₆N₆的表面修饰、粒径控制以及与其他材料结合的方法,以提升其气体吸附和传感性能。
总结
态密度(DOS)作为连接电子结构与催化性能的核心桥梁,在电催化研究中发挥着不可替代的作用,其通过量化电子能级分布,为解析活性起源、指导材料设计提供了原子级视角,同时在计算方法与应用领域的创新推动下,不断拓展其理论深度与实用价值。
从理论意义来看,DOS不仅能揭示活性位点的关键轨道,还能通过d带中心、DOS等参数建立“电子结构–吸附能–反应活性”的关联。
在计算方法上,态密度的发展趋势聚焦于机器学习加速与原位动态模拟:机器学习通过提取DOS特征训练预测模型,将催化剂筛选效率提升1-2个数量级。
当前面临的挑战主要集中在强关联体系的DOS计算,传统DFT难以准确描述d电子的强关联效应,导致态密度峰值位置偏差,需发展多体方法以提升精度。
通过深入理解DOS的理论基础与顶刊应用,研究者可精准设计催化剂的电子结构——如通过掺杂调控d带中心、引入缺陷,实现对吸附能与反应能垒的定向优化。
未来,随着计算方法的完善与原位表征技术的进步,DOS分析将在“催化剂基因工程”中发挥核心作用,推动能源转化技术从经验开发迈向理性设计,为燃料电池、电解水等领域的高效催化剂开发提供坚实的理论支撑。