不同泛函计算对能带结构的影响是密度泛函理论(DFT)研究中的核心问题之一。通过比较不同泛函(如LDA、GGA、HSE06、PBE+U等)在计算材料能带结构时的表现,可以揭示这些泛函在描述电子结构、能隙、带隙、态密度(DOS)等方面的能力差异。
不同泛函的基本概念与特点
LDA(Local Density Approximation)
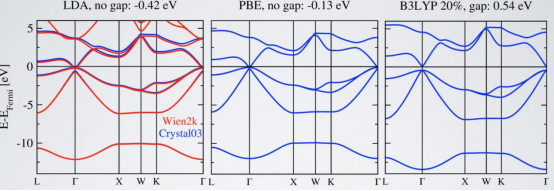
LDA是一种最基础的交换–关联泛函,其核心假设是交换–关联能仅依赖于局部电子密度。尽管LDA在某些情况下表现良好,但在处理复杂电子结构时,其局限性明显。例如,LDA通常低估了材料的能隙,导致计算出的能带结构与实验结果存在较大偏差。

GGA(Generalized Gradient Approximation)
GGA在LDA的基础上引入了梯度项,能够更准确地描述电子密度的空间变化。Perdew等人提出的PBE泛函是GGA的典型代表,广泛应用于金属、半导体和绝缘体的电子结构计算。然而,PBE泛函在某些情况下仍会低估能隙,尤其是在处理强关联体系时,其表现不如更高阶的泛函。
杂化泛函(Hybrid Functionals)
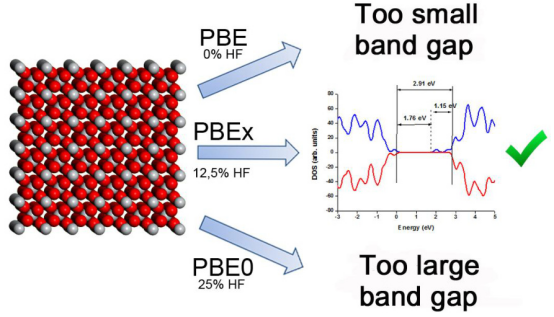
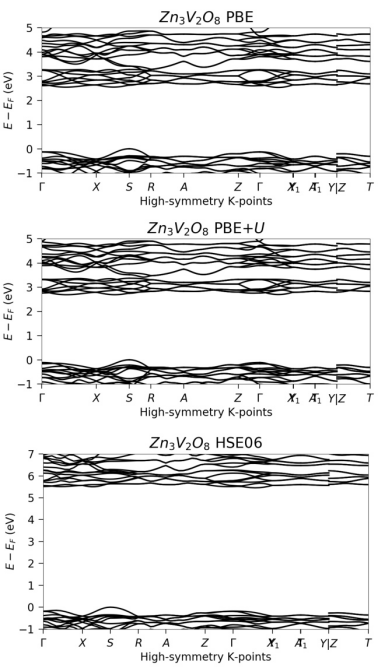
杂化泛函结合了Hartree-Fock(HF)交换和DFT中的交换–关联泛函,能够更准确地描述电子相互作用。例如,HSE06是一种常用的杂化泛函,它通过引入屏蔽库仑势来近似HF交换,从而提高计算精度。此外,PBE+U泛函通过引入强排挫能(U)来修正强关联体系中的电子结构,特别适用于过渡金属氧化物等材料。
GW+DMFT方法
GW+DMFT是一种结合了GW近似和动力学平均场理论(DMFT)的方法,用于处理强关联电子体系。这种方法在计算能带结构和态密度时表现出更高的精度,尤其是在处理复杂电子结构和磁性材料时。
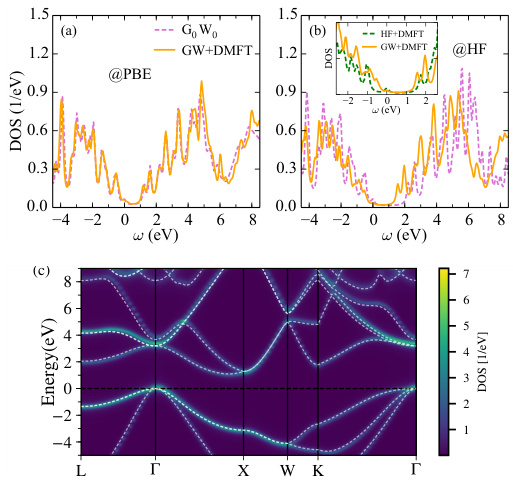
不同泛函对能带结构的影响
能隙的预测
不同泛函对材料能隙的预测结果存在显著差异。例如,在Zn3V2O8材料中,使用PBE泛函计算的能隙约为1.2eV,而使用HSE06泛函计算的能隙则为2.8eV,与实验值更为接近。这表明,PBE泛函在某些情况下低估了能隙,而HSE06泛函则提供了更准确的结果。类似地,在β-SnO材料中,DFT-D3-BJ泛函计算的能隙约为1.5eV,而HSE06泛函计算的能隙则为2.0eV,显示出HSE06在描述能隙方面的能力更强。
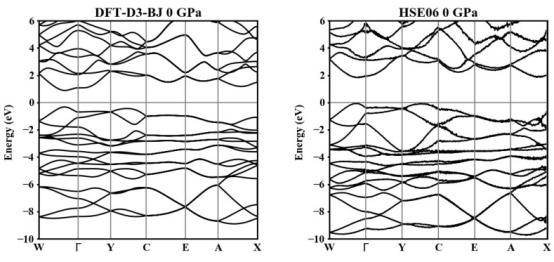
能带结构的细节
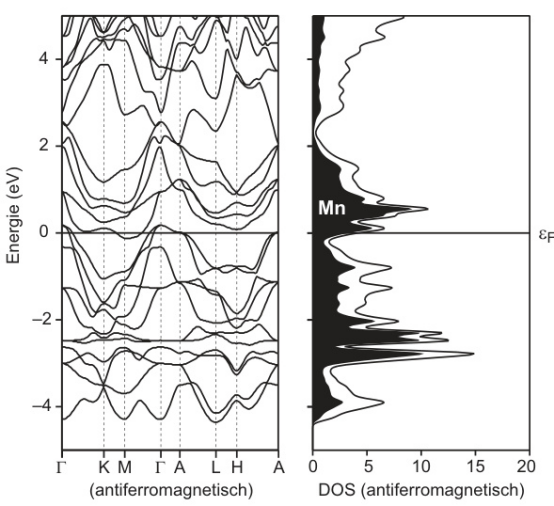
不同泛函对能带结构的细节也有显著影响。例如,在Mn基材料中,使用PBE泛函计算的能带结构较为平坦,而使用HSE06泛函计算的能带结构则显示出更多的能带交叉和更复杂的电子态分布。这表明,HSE06泛函能够更准确地描述材料的电子结构,尤其是在处理复杂能带结构时。
态密度(DOS)的计算
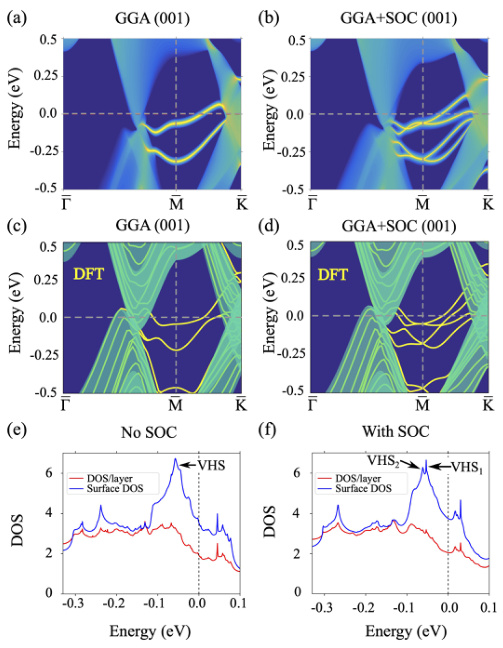
态密度(DOS)是描述材料电子结构的重要参数,不同泛函对DOS的计算结果也存在差异。例如,在二维材料Pt2HgSe3中,使用GGA方法计算的DOS显示出较高的峰谷密度,而使用GGA+SOC方法计算的DOS则显示出更平滑的分布。这表明,自旋轨道耦合(SOC)对DOS的计算结果有重要影响,尤其是在处理具有复杂电子结构的材料时。