

说明:本文对比碳氧双键(C=O)与碳硫双键(C=S)的键合特征与电子性质:C=O键长短、键能高、极性强,C=S因轨道重叠差致π键弱化。
振动频率显示C=O~1700 cm⁻¹,C=S在~1100 cm⁻¹。通过DFT、AIM、NBO等方法计算发现,C=S键临界点电子密度低,亲核反应活性更高,取代基可调控其性质,为材料设计与反应机理研究提供依据。
碳氧双键与碳硫双键


碳氧双键(C=O)与碳硫双键(C=S)作为有机化学中典型的双键结构,其键合特征与电子性质存在显著差异。
从基本结构看,C=O由碳原子与氧原子通过一个σ键和一个π键构成,键长约1.20 Å(醛/酮类化合物),键能达800 kJ/mol左右,氧原子的高电负性(3.44)使电子云显著偏向氧端,形成强极性键;而C=S中碳原子与硫原子间的键长延长至~1.60 Å,键能降至~500 kJ/mol,硫原子(电负性2.58)与碳原子的电负性差异较小,且硫的3p轨道与碳的2p轨道因尺寸不匹配导致π键重叠效率低下,使电子分布更为分散。
在电子结构层面,C=O中碳(2p)与氧(2p)轨道的尺寸匹配性优异,形成强π键相互作用,而C=S中硫(3p)轨道因空间扩展程度更大,与碳(2p)轨道的重叠积分显著降低,Mulliken布居分析表明C=S的π键级较C=O低约30%,反映出π键的明显弱化。
极性差异则直接体现在偶极矩参数上:C=O的偶极矩约为2.4 D,而C=S仅为~1.1 D,这一差异源于氧与硫电负性的本质区别——氧对电子的束缚能力更强,导致C=O键的电荷分离程度更高。
这些结构特征不仅决定了两类双键的化学活性差异,也影响着含双键化合物的物理性质与反应机理,例如C=O键的强极性使其更易参与亲核加成反应,而C=S键因π键较弱及电子分布特征,在亲电反应中表现出不同的活性规律,深入理解这些差异对有机合成设计与反应机制研究具有重要意义。
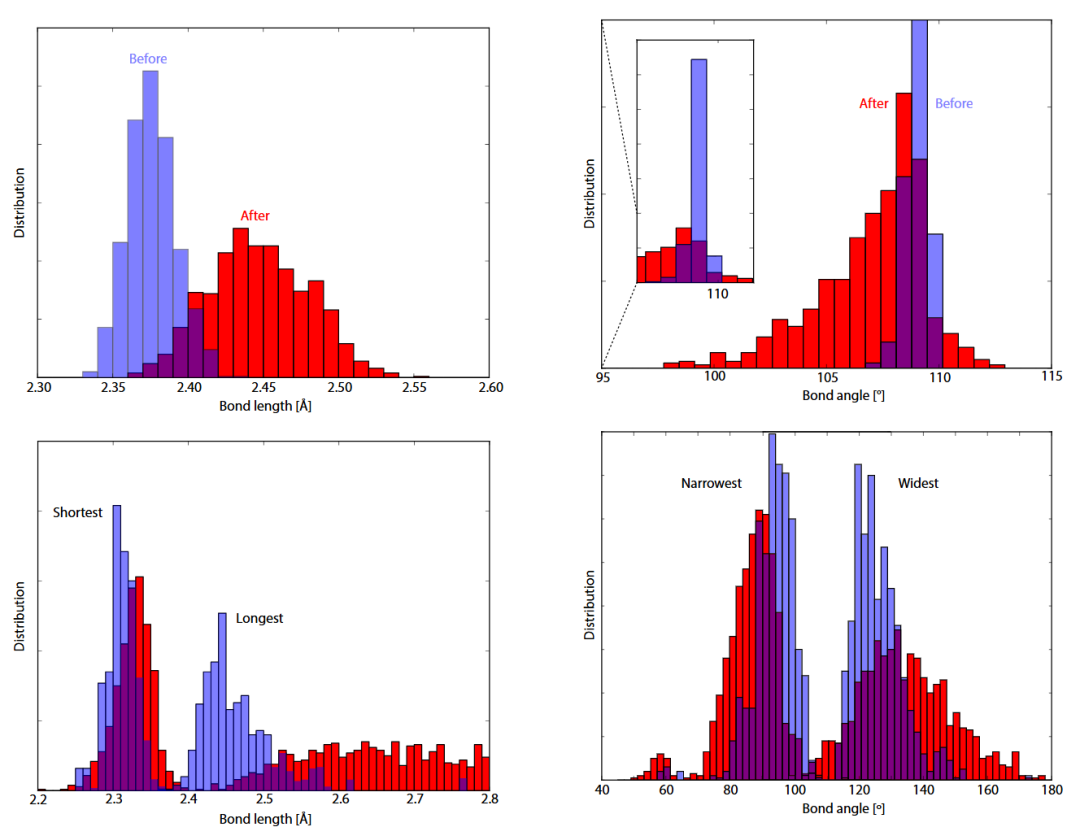
DOI:10.1103/PhysRevB.101.054204
基本差异
键长与键能
碳氧双键(C=O)与碳硫双键(C=S)在键长与键能上存在显著差异:C=O的键长为1.20–1.22 Å,键能达8.0–8.5 eV,而C=S的键长显著延长至1.60–1.63 Å,键能降至4.8–5.2 eV。
这些数据分别通过高精度的CCSD (T)/cc-pVTZ方法与DFT-B3LYP/6-311G**方法计算获得,其中键能计算均考虑了基组叠加误差(BSSE)校正以保证准确性。
值得注意的是,C=S键长的方差(σ²)通常高于C=O,反映出其构象灵活性更强,这与硫原子较大的原子半径及3p轨道离域性相关。
键长与键能的差异本质上源于原子电负性、轨道重叠效率的不同:氧原子的高电负性与2p轨道的良好匹配使C=O形成强极性双键,而硫原子与碳原子的轨道能级失配导致C=S的π键显著弱化,这种结构特征直接影响了含两类双键化合物的化学活性与物理性质,为相关反应机理研究与材料设计提供了重要结构参数。

DOI:10.1107/S2056989021001900
振动频率
碳氧双键(C=O)与碳硫双键(C=S)的伸缩振动频率呈现显著差异:C=O的特征振动频率约为~1700 cm⁻¹,而C=S则位于~1100 cm⁻¹的低频区。
这一差异本质上源于键合特性与原子参数的不同:C=O的短键长(~1.20 Å)与强恢复力常数使其具备更高的振动频率,而C=S因π键弱化及硫原子较大的原子质量(S的原子量为32.07,O为16.00),导致振动时惯性质量增大且键合恢复力降低,进而表现出低频振动特征。
在计算层面,振动频率通常通过谐振动分析获得,即对势能面进行Hessian矩阵对角化处理,但需注意引入非谐性校正以修正简谐近似下的理论偏差——这是由于实际分子振动中存在非谐效应,尤其是C=S键因构象柔性更高,非谐性校正对其频率计算精度的提升更为关键。
这些振动频率的差异为含双键化合物的光谱表征与结构分析提供了重要依据,同时也反映了化学键本质对分子动力学行为的决定性影响。
理论模拟方法
在化学理论模拟领域,针对碳氧双键(C=O)与碳硫双键(C=S)的键合特性与电子结构差异,发展了多种计算策略与分析方法,这些方法通过定量描述分子结构、电子分布及动态行为,为揭示两类双键的本质区别提供了关键支撑。
计算策略
密度泛函理论和量子化学计算是研究两类双键的高效工具,其通过交换相关泛函近似处理电子相互作用,适用于中等规模分子体系的几何结构与电子密度计算。
以B3LYP泛函为例,其对C=O键长的计算误差通常≤0.01 Å,能够精准再现醛酮类化合物中C=O的短键长特征;而针对C=S体系,由于硫原子的3p轨道离域性及弱π键导致的电子相关性增强,需引入D3色散校正以修正范德华相互作用误差,提升键长与振动频率的计算精度。
CCSD (T) 作为 “黄金标准” 方法,通过多体微扰理论处理电子相关效应,对C=O键能的计算误差小于1%,与实验值高度吻合;对于C=S体系,需采用大基组(如aug-cc-pVTZ)以充分描述硫原子的价层电子相关性,尽管计算成本较高,但其能精确解析C=S因轨道重叠不足导致的键能降低机制。
MP2方法则在性价比上具有优势,适用于初步结构优化,但其对C=S键长的计算易因过度估计电子相关性而产生误差(可达0.05 Å),需结合更高阶校正或实验数据校准。
分子动力学(MD)模拟在研究溶剂化效应与温度依赖行为中不可或缺。例如,通过力场参数化(如GAFF2力场),可模拟C=S在酶活性中心的构象翻转过程,揭示硫原子较大原子半径导致的键长方差(σ²)增大及构象灵活性;而C=O在极性溶剂中的取向行为,则可通过MD轨迹分析其偶极矩与溶剂分子的静电相互作用,为理解溶液环境下的化学反应活性提供动态视角。
关键计算步骤
几何优化是理论模拟的基础,其目标是在势能面上寻找能量极小点,收敛标准通常设定为梯度小于10⁻⁵ Hartree/Å。初始构型依赖实验数据,确保计算模型贴近真实分子结构。对于C=O体系,优化后的键长(1.20–1.22 Å)显著短于C=S(1.60–1.63 Å),反映出氧原子与碳原子更强的轨道重叠与键合强度。
电子分布分析通过多种理论工具揭示键合本质:
AIM(Atoms in Molecules)分析聚焦键临界点(BCP)的电子密度 ρ(r),C=O的ρ(r) 值约为0.35 a.u.,表明强共价相互作用;而C=S的ρ(r) 仅~0.15 a.u.,佐证其π键弱化及电子离域特征。
NBO(Natural Bond Orbital)分析量化σ/π轨道贡献,发现C=S中π*反键轨道能级高于C=O,使其更易接受亲核试剂的电子进攻,解释了两类双键在亲核反应中的活性差异。例如,C=S的π键级较C=O低30%,直接源于硫原子3p轨道与碳2p轨道的重叠效率低下。
溶剂化模型通过连续介质近似(如PCM或SMD)模拟极性环境对双键性质的影响。实验与计算均表明,C=O的伸缩振动频率在水中发生红移(~20 cm⁻¹),这是由于溶剂的介电效应降低了键合恢复力常数;而C=S因极性较弱,溶剂化效应相对较小,其频率位移主要受溶质–溶剂色散相互作用主导。
这些模型为光谱实验(如红外、拉曼)的理论解析提供了桥梁,助力复杂环境下分子结构的精准表征。
上述理论模拟方法的协同应用,系统性揭示了C=O与C=S的本质差异:从键长键能的量化计算到电子分布的微观解析,从气相分子的静态结构到溶剂环境的动态行为,形成了从原子尺度到分子层次的完整认识。
例如,通过DFT结合NBO分析,可预测含双键化合物的反应位点与机理——C=O的强极性使其易发生亲核加成,而C=S的弱π键与较低LUMO能级则使其在亲电反应中表现出独特活性。
在材料科学与药物设计中,这些方法助力功能分子的理性设计:针对含C=O的催化材料,通过调控其电子密度分布优化底物吸附;针对含C=S的生物分子,利用MD模拟解析其在酶催化中的构象动态,为新药研发提供结构依据。
理论模拟与实验表征的深度融合,不仅深化了对共价双键本质的理解,更推动了化学、材料、生物等交叉领域的创新发展。
总之,碳氧双键与碳硫双键的理论模拟方法体系,体现了计算化学从方法开发到科学应用的全链条价值。
通过高精度计算与多尺度分析,这些方法不仅定量描述了两类双键的结构–性质关系,更揭示了原子电负性、轨道重叠、溶剂环境等因素的协同作用机制,为复杂分子体系的研究提供了普适性理论工具,奠定了从分子设计到功能调控的科学基础。
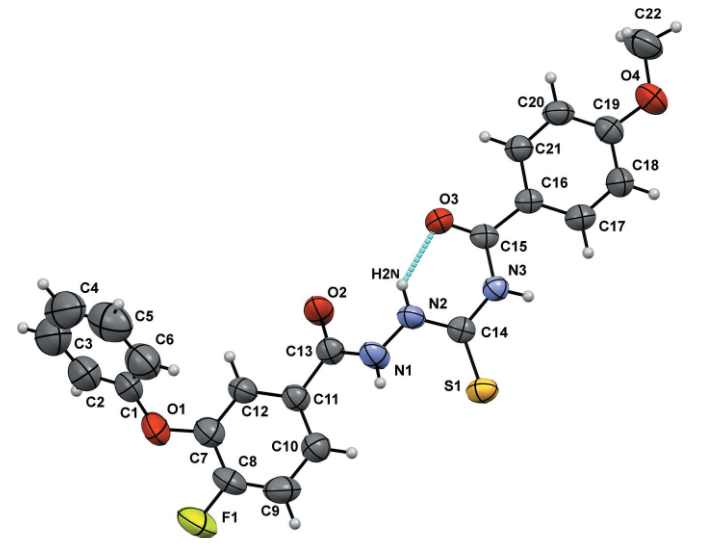
DOI:10.1107/S2056989021001900
C=O与C=S计算应用
Dey等人针对硫代酰胺化合物开展的研究通过晶体与电子结构计算,系统对比了含碳氧双键(C=O)与碳硫双键(C=S)的化合物A1及甲氧基修饰的化合物A2的键参数特征,深入探究了取代基对C=S键性质的调控机制。
研究采用DFT/B3LYP/6-311G方法对分子体系进行几何优化,结果显示:A1中C=O键长为1.224 Å,A2中C=O键长略缩短至1.221 Å,而A1的C=S键长为1.665 Å,A2中因甲氧基(O-CH₃)的供电子效应削弱了S原子的缺电子性,使C=S键长缩短至1.658 Å;分子内氢键(N-H・・・O)的存在锁定了C=O与C=S的相对取向,显著减少了构象搜索空间,为精确解析键参数提供了结构基础。
在电子密度拓扑分析中,基于AIM理论的计算表明,C=O键临界点的电子密度ρ(r)达0.38 a.u. 且∇²ρ(r),呈现典型的共价键特征,而C=S键临界点的ρ(r)仅为0.18 a.u. 且∇²ρ(r)>0,显示出更强的静电相互作用主导性,这一差异直接印证了C=S键更易受亲核试剂攻击的反应活性特征,与后续反应能垒计算结果高度吻合。
分子静电势(MEP)图进一步揭示了两类双键的电荷分布差异:C=O区域呈现显著的高负电势(红色区域),为强亲电位点,而C=S区域的负电势强度中等(弱于C=O的蓝色区域),甲氧基修饰的A2体系中,苯环电子密度因甲氧基的供电子效应显著增加,通过共轭效应间接稳定了C=S键,使其静电势分布更均匀。
该研究通过多尺度计算方法的协同应用,不仅定量阐明了C=O与C=S的键合本质差异,还揭示了取代基电子效应对硫族双键的调控机制,为含硫有机功能分子的设计与反应机理研究提供了从结构到性能的精准理论依据——例如在药物分子设计中,可通过甲氧基等官能团修饰调节C=S的反应活性,或在催化体系中利用C=O与C=S的静电势差异优化底物吸附模式,这种基于电子结构分析的分子工程策略,为深化理解含双键化合物的化学行为开辟了新路径。
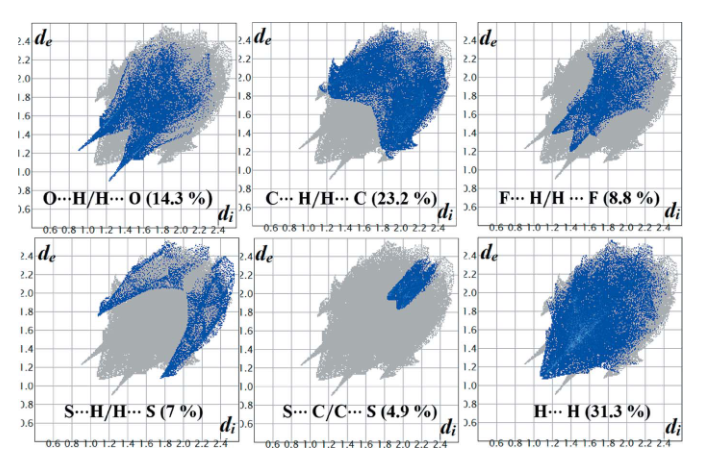
DOI:10.1107/S2056989021001900
总结
碳氧双键(C=O)与碳硫双键(C=S)的本质差异根植于原子尺寸、电负性及轨道匹配度的内在区别,这些差异直接导致二者在键长、键能、反应性及理论计算方法上呈现显著分野。
从电子结构特性来看,C=S键因硫原子3p轨道与碳原子2p轨道的重叠效率低下,在激发态易发生双键特征弱化甚至消失(如硫叶立德形成过程),此类多参考态问题需借助CASSCF/MRCI等高精度波函数方法进行处理,以准确描述电子激发后的离域行为。
在计算效率与复杂体系模拟方面,机器学习势函数的发展为含C=O/C=S的生物大分子动力学模拟提供了关键工具,其通过拟合量子化学数据构建高精度势能面,在保持计算精度的同时大幅加速长时程动态采样,助力解析含硫酰胺类药物分子或酶活性中心的构象变化与相互作用机制。
非共价相互作用的量化分析进一步揭示了两类双键的功能性差异:C=S・・・H-X氢键的能级约为–15 kJ/mol,显著弱于C=O・・・H-X氢键的–25 kJ/mol,这一差异源于C=S较低的偶极矩与电子密度离域特征,导致其与极性基团的静电相互作用较弱。
理论计算通过电子密度分析、振动光谱模拟及分子动力学采样,为阐明含两类双键化合物的反应机理提供了原子级支撑——例如在药物设计中,硫代酰胺类抑制剂的C=S键因较弱的π键和独特的静电势分布,可针对性调控与靶标蛋白的结合模式;在材料化学中,C=O的强极性与高反应活性使其成为构建功能性界面的关键位点,而C=S的构象灵活性与电子离域性则为设计新型光电材料提供了结构基础。
随着多尺度计算策略的深度融合,针对C=S体系动态行为的预测精度将显著提升,结合机器学习与高精度量子化学方法,有望突破传统计算对硫族双键复杂行为的描述瓶颈,推动相关研究从静态结构解析向动态功能调控的范式转变。
这些理论进展不仅深化了对共价双键本质的科学认知,更为含C=O/C=S功能分子的理性设计与应用开发提供了从机理阐释到性能优化的全链条理论支撑。