在异相催化中,活性位点指的是催化剂表面实际参与反应的原子或原子群,是发生催化反应的具体位置。
早在1925年,Taylor就提出催化剂表面只有一小部分原子(通常位于拐角、棱边等晶格缺陷处)具有催化活性。换言之,并非催化剂表面的每个原子都能催化反应,只有特定构型的一小部分原子/原子团构成了活性中心。
活性位点在催化反应中起决定性作用——反应物在此处吸附、活化并转化为产物。活性位点的性质(如结构、价态、电子性质等)直接影响反应路径、能垒和速率,从而决定催化剂的活性和选择性。
识别和理解真实的活性位点对于理性设计高效催化剂至关重要,但由于催化过程中活性物种的动态变化及表征技术的局限性,准确确定活性位点一直是实验和理论上的挑战。
DFT作为一种量子化学计算方法,可以提供催化剂表面吸附、反应的热力学和电子结构信息。通过DFT,可以比较不同位点的反应性能,从而推断哪个位点是主要的活性中心。以下是DFT确定活性位点的几个常用思路:
吸附能(或称吸附自由能)计算是判定活性位最直接的方法之一。它衡量反应物或中间体在催化剂不同候选位上的结合强度。
一般而言,有效的活性位点应当能够适度地吸附关键反应物或中间体:既不能过弱(否则无法活化反应物),也不能过强(否则中间体难以脱附,反应停滞),这体现了Sabatier原理。
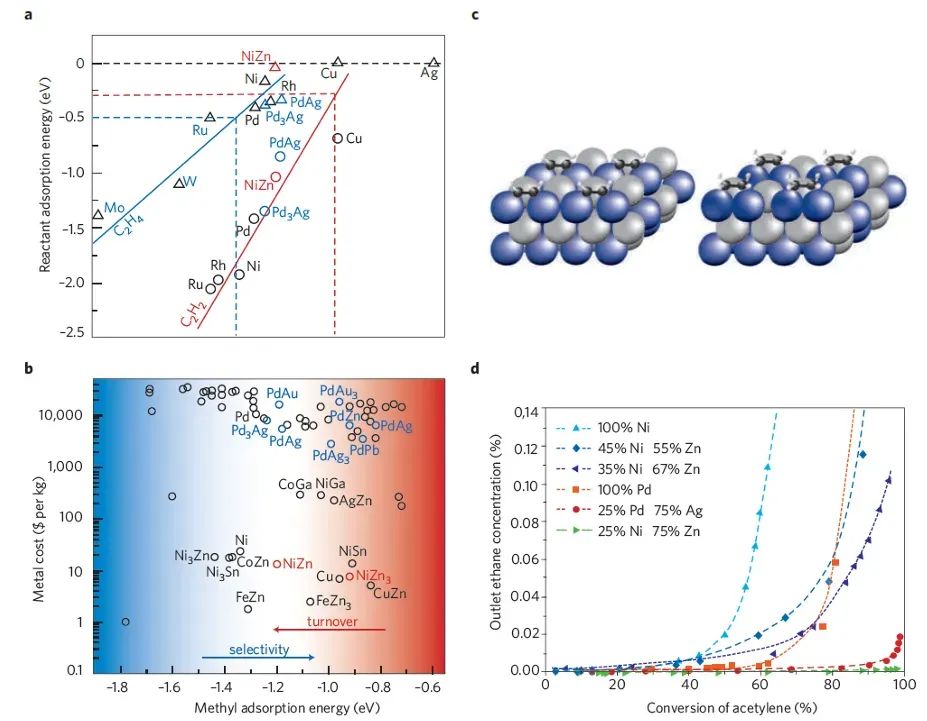
通过DFT计算多个可能位点的吸附能,可筛选出“适中”吸附的位点作为潜在活性中心。例如,在金属合金催化剂中,DFT模拟发现反应物更倾向于吸附在特定金属原子上,而非另一金属上,从而指示前者为活性位。
在Ni-Zn合金催化加氢反应的研究中,DFT计算约70种合金表面的中间物CH*吸附,结果显示吸附物优先与Ni原子结合而非Zn,这意味着Ni是主要承担反应的活性位,而Zn则主要通过改变Ni的电子性质来助催。
DIO:
10.3390/catal8100478
再如,对于氧还原反应(ORR)的非贵金属单原子催化剂Fe-N-C体系,理论和实验均发现Fe-N_x位点虽能吸附O₂等中间物,但往往吸附过强,类似于Pt催化剂对氧中间体的过强结合,导致生成物难以脱附,从而限制了反应动力学。这提示单一Fe-N_x位的结合力偏强,不利于ORR快速进行,需要通过调控其电子结构来降低吸附强度。
DFT计算的吸附能还能用于构建活性火山曲线。通过改变催化剂位点的电子/结构特征,吸附能会变化,从而影响活性。理想活性位对应于火山曲线顶点的吸附强度,一个案例是对FeN₄位点引入轴向配位杂原子。
除了静态吸附能,比对完整反应路径上的能量垒更能直接体现哪个位点反应更容易进行。具体做法是:在不同候选位点上,计算反应各步的反应能垒(过渡态能)或反应自由能变化,比较决定步骤(速控步)的能垒或自由能高低。
活性位点应呈现最低的反应能垒或最小的限制步骤自由能增量。对于电催化反应,常使用Nørskov提出的可计算氢电极(CHE)模型,通过DFT获得各中间体的自由能并加上电极电位修正,得到每一步反应的自由能变化ΔG;其中最大的正ΔG对应的步骤即为速控步骤,其数值决定了催化所需的过电位。
过电位η可以通过η = |ΔGmax|/e − 1.23V(以ORR四电子还原的1.23V平衡电位为基准)计算得到。
因此,在不同位点上,计算所得最大ΔG(或最低电位下所需克服的ΔG)越小,该位点所需的驱动力(过电位)就越低,即催化活性越高。
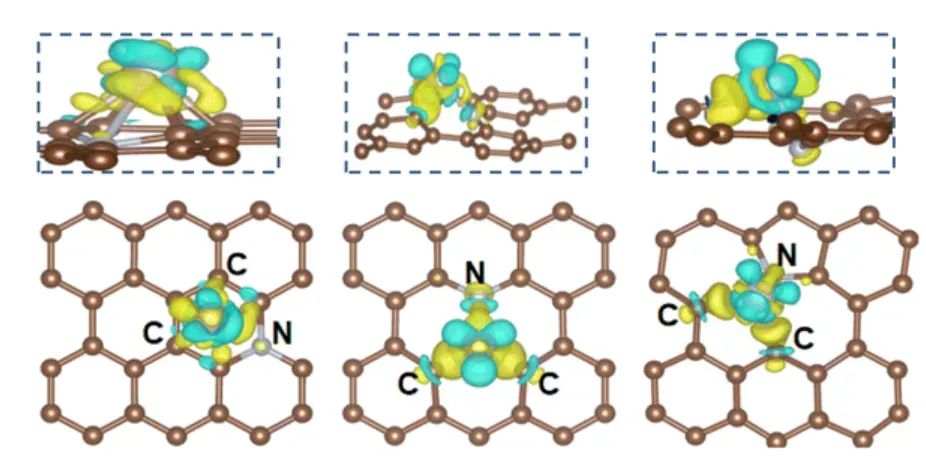
差分电荷密度图(也称电荷密度差分)是DFT分析活性位点化学作用的一种直观手段。通过将吸附前后的电子密度作差,差分电荷密度图以等值面形式显示电子的积累与缺失区域(常用黄色/红色表示电子富集,蓝色/绿色表示电子缺失)。
这可以帮助识别在吸附过程中电子从哪里转移到了哪里,也即哪一个原子在向吸附种提供电子或接受电子,从而判断哪个原子在键合作用中起主要作用。
Bader电荷分析是另一种量化各原子电荷状态的方法。通过将体系总电子密度按照零通量面分区,Bader分析可以给出每个原子的净电荷(相对价态的电子得失情况)。在催化中,活性位原子的Bader电荷常用于判断其氧化还原状态以及与载体或反应物的电荷转移关系。
DFT还能通过分析电子能态分布,提供活性位电子结构与反应性的关联信息。
其中最常用的d带中心理论,由Nørskov等提出,已成为金属催化活性的重要描述符。
简言之,过渡金属表面的d电子能带相对于费米能级的位置(即d带中心的高低),影响着其与吸附种的键合作用强度:d带中心越接近费米能级,金属与吸附种轨道重叠越多、反键轨道填充更少,通常意味着更强的化学吸附;反之,d带中心越远离费米级(越低),与吸附物的相互作用越弱。
当比较不同活性位或调变催化剂时,d带中心可以用作预测吸附强度乃至活性的指示器。
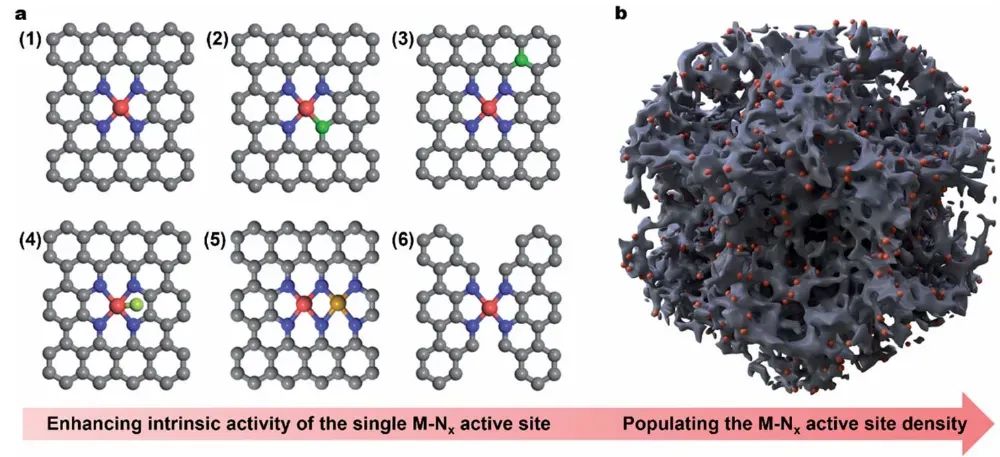
对于单原子催化剂,其d带中心亦受到配位环境的调制。DFT的投影态密度(Projected DOS, PDOS)计算常用于获取活性原子d带中心值及轨道分布。
Fe-N-C单原子催化剂ORR活性位的判定
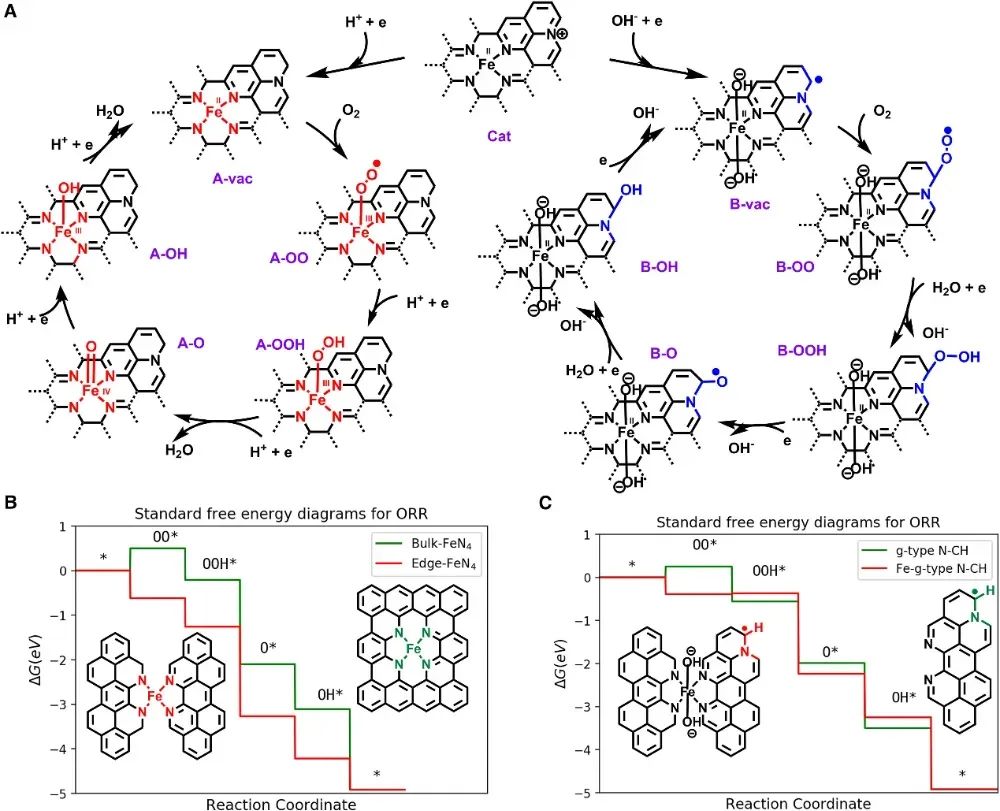
Chen等人在2020年的研究中采用Gaussian 09量子化学程序,在B3LYP泛函水平对模型簇进行计算。
他们分别构建了代表Fe-N-C材料中两种候选活性结构的簇模型:一种是FeN₄(Fe以四个吡啶氮配位,嵌于碳骨架中),另一种是g型N-CH(石墨化N原子邻位带有氢的碳,模拟Fe缺位但存在N的碳缺陷位)。
考虑到酸碱条件的差异,计算特别比较了H⁺和OH⁻在这两种位点上的吸附行为和ORR中间体反应。
DFT结果预言,在酸性介质中,FeN₄位点对O₂的吸附和O–O键断裂最为有利,是ORR的活性位;然而在碱性介质下,FeN₄位上的反应遇到了困难(例如H⁺难以在FeN₄上吸附,计算得到H⁺在FeN₄上的吸附自由能高达+1.16eV,极其不利),这与实验观察到Fe-N-C在碱性溶液中活性下降相符。
相反,DFT计算显示石墨化N-CH位在碱性条件下能够更有效地参与ORR:该位点可以接受OH⁻或参与质子转移,从而补偿FeN₄位的不足。因此作者提出双活性位模型:在酸性溶液中Fe-N₄是主要活性位,而在碱性条件下邻近N的碳基位(g-N-CH)成为主导活性位。
为验证上述理论,作者精心合成了一种同时含有FeN₄和N-C缺陷位的Fe单原子催化剂,并通过电化学测试发现该材料在酸、碱两种介质中均表现出优异且稳定的ORR活性。这证明两个位点在各自条件下协同作用,实现了宽pH范围的高效催化。
通过DFT计算与实验结果的结合,该工作清晰地指认了Fe-N-C材料中真实的活性中心:FeN₄负责酸性ORR,石墨化N-CH位负责碱性ORR。这一发现澄清了长期争论的Fe-N-C催化剂活性位问题,提示在设计此类SAC时需针对应用环境考虑不同活性位,并为今后开发pH通用的ORR催化剂提供了思路。
DOI:
10.1016/j.xcrp.2020.100115
Yang等人在2025年的研究中系统地考察了上述不同N配位下Fe和Ru SAC的HER活性。他们在DFT计算中构建石墨烯簇/片模型,引入N缺陷并放置单个Fe或Ru,然后计算H吸附自由能ΔG(H)、HER反应自由能图以及相应的电子性质(包括Bader电荷、金属d带中心等)。并通过计算各种构型下的结合能评估单原子的锚定稳定性。还使用火山图分析将HER的交换流密度或过电位与ΔG(H*)进行关联。
计算结果表明,Fe和Ru在不同配位环境中表现出明显不同的行为:配位数越少(如Fe-N₂,Ru-N₁),金属d电子填充越高、与载体作用越强,因而H吸附ΔG偏负,有利于H原子结合,但若过强则可能不利于H₂脱附;配位数越多(Fe-N₄,Ru-N₄),则金属偏正电、d态下移,H吸附ΔG偏正,吸附过弱不利于H生成。综合来看,中等配位的缺陷(如Fe-N₂、Ru-N₁–₂)能够将ΔG(H*)调控在接近0的最佳范围,从而在火山图上位于顶点附近。具体而言,他们发现Fe-N₂和Ru-吡咯N₁位点分别是两种金属的最优构型:两者计算的HER过电位低至约0.23 V和0.26 V,显著优于传统的Fe-N₄或Ru-N₄位点。与此同时,Fe-N₂和Ru-N₁尽管配位较低但通过与载体强相互作用仍保持了一定的稳定性。Bader电荷分析进一步揭示了原因:在这些优活性构型中,Fe或Ru原子带有适中的正电荷,表明与周围N/C原子有合理的电荷转移。这种电荷状态增强了金属-载体的电子耦合,使得H既能被吸附又能被释放(未过度稳定氢负离子),从而促进了HER双原子氢形成步骤。相比之下,在Fe-N₄或Ru-N₄位点,金属电荷更高(更缺电子)导致H过度稳定无法高效脱附,或者在极端情况下金属电荷过低导致H⁺难以结合,这与其较差的ΔG(H*)和较高过电位一致。该研究结论是:通过调控单原子周围的配位原子数目和类型,可以优化活性位的电子结构,从而实现对催化活性的调控。对于HER,适度的配位缺陷(如N₂或N₁类型)为单原子提供了最佳的电子环境,使其对反应中间体H的亲和力恰到好处,体现为ΔG(H)≈0和极低的过电位。这一发现为单原子催化剂的设计提供了重要指导:在保证单原子稳定性的前提下,引入配位缺陷或异质配位,可激发单原子的活性。例如,可通过合成前驱体或后处理方法创造缺位或低配位环境来提高SAC的活性。更广泛地,该工作亦验证了DFT计算预测与火山关系在单原子催化领域的适用性,即理论上存在一组合适的电子结构指标(如金属正电荷和键能)使单原子位达到性能峰值。这对于理性设计非贵金属高效电催化剂(如Fe、Ru基SAC替代Pt催化HER)具有重要意义。
通过以上探讨可以看出,密度泛函理论计算已成为探明单原子催化剂和电催化体系活性位点的强大工具。
DFT能够从原子和电子层面揭示:哪个位点吸附反应物/中间体最优(吸附能分析)、哪个位点使反应能垒最低(路径和过渡态分析)、活性位的电子转移特征(差分电荷密度、Bader电荷)、以及活性位的电子结构与反应性的内在关系(d带中心、态密度等)。
随着DFT方法学和计算能力的进步(例如更精确的功能、含溶剂和电场的模型等)以及与机器学习等手段的结合,理论计算将在活性位点识别与催化剂设计中发挥更为核心的作用,推动催化科学朝着定量可预测和可定制设计的方向发展。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!