通过金属、半导体与二维材料的具体案例,展示了稳定性评估在不同材料体系中的应用价值,进一步强调了理论计算在解释实验现象(如相变、结构稳定性变化等)中的重要作用,最终指出理论与实验协同已成为现代材料研究中不可或缺的趋势。
材料的稳定性通常指在特定条件(温度、压力、化学环境等)下保持结构不发生自发分解或转变的能力。一般可分为热力学稳定性、动力学稳定性以及环境/热稳定性等类型。
热力学稳定性:指体系处于最低自由能态的程度。若一种材料的化学计量和结构在给定条件下对应的自由能最低,则热力学上稳定。常用形成能(或称生成能)评估。
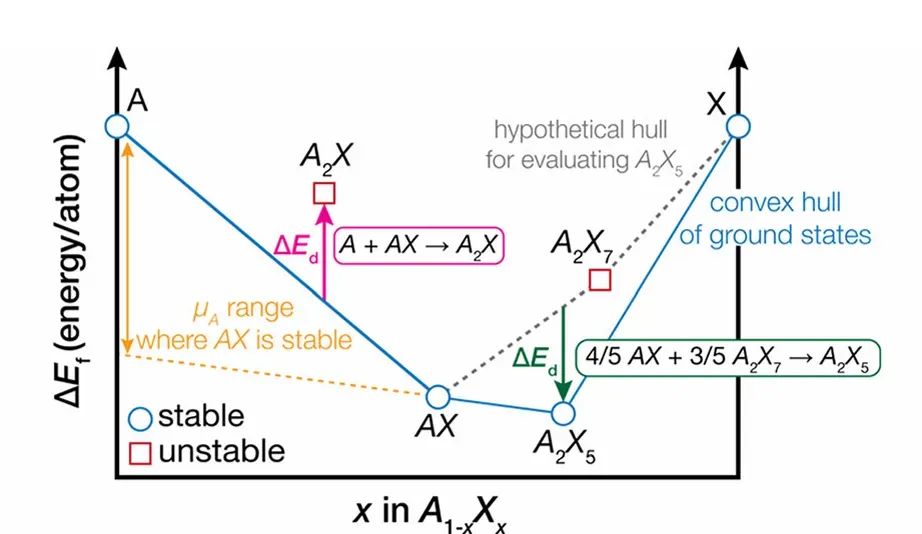
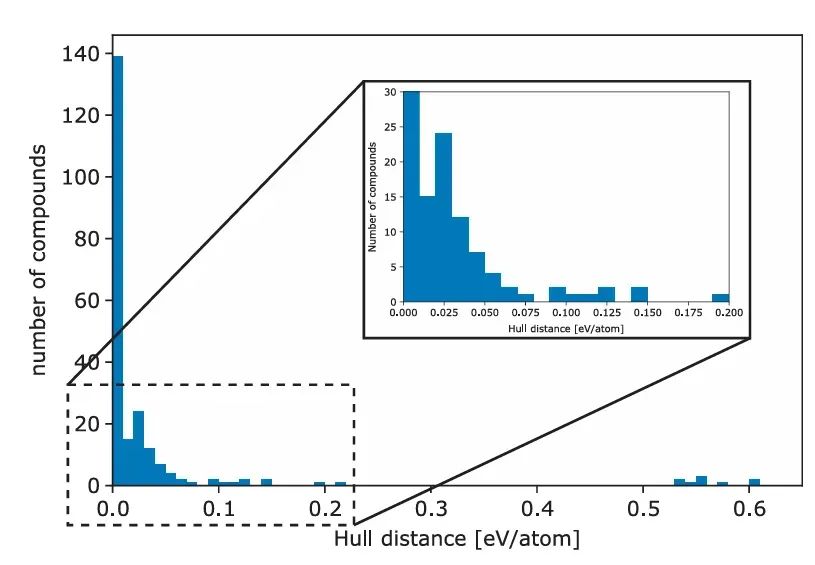
例如,在多组元合金或化合物体系中,可以构造能量-成分凸包图:位于凸包上的相点(图中蓝色圆圈)表示热力学稳定,相对于任何可能的分解反应都具有最低能量;高于凸包的结构则可自发分解成凸包上的邻近平衡相。
因此,材料与元素或更简单化合物组合的形成能越低,越有利于形成和稳定。类比地,结合能(或内聚能)是指将材料中各原子分离到孤立原子的能量差,也是热力学稳定性的度量之一。
对热力学稳定性的计算常在零温下进行,但也可通过包括声子自由能等方式扩展到有限温度。
DOI: 10.1007/s10853-022-06915-4
动力学稳定性:描述材料从一种相态向另一种相态转变时所需克服的势垒高低。即使某相在热力学上不是最低能量(如图中凸包上方的点),只要向更稳定相的转变需要较高的激活能垒,该相仍可长期存在。例如,钻石(C)在常温下虽非热力学最低相(石墨自由能更低),但由石墨转变为钻石需要极高的能垒,所以钻石在常温下表现为动力学稳定。理论上可计算相变路径和过渡态能垒(如弹性带算法等),也可根据有限温度下的自由能竞争来定性分析何时由动力学主导。然而,由于实际合成往往受复杂条件影响,动力学稳定性难以仅靠第一性原理准确预测,只能通过对比不同相的能垒和微观机制进行分析。热稳定性与环境稳定性:指材料在高温或特定环境(氧化性气氛、湿度、化学试剂等)下保持结构完整的能力。热稳定性可以通过计算分解能(或材料熔融温度)的变化来估计,实验上则用热重分析(TGA)等方法测定降解温度。理论上常结合原位高温模拟(如AIMD模拟高温结构演化)来评估。环境稳定性关注材料在常温或特定化学环境中的抗氧化、抗腐蚀性,例如某些半导体材料(如黑磷)易在空气中氧化而失活,这可通过计算氧化反应的形成能和路径来分析。总之,热力学稳定性强调能量最低原则,动力学稳定性强调能垒阻止相变,两者决定相变能否发生;环境/热稳定性则涉及额外的温度或化学反应因素。从计算角度看,前者多用总能量和自由能比较,后者则辅以动力学模拟和反应能分析。
形成能(formation energy)是评估热力学稳定性的基础量:它定义为材料的总能与其组元元素在参考态(通常为纯金属或分子气体)总能之差。形成能越低,表明该相相对于元素更稳定。计算形成能时,需选择合适的元素化合态基准,并进行化学势严格归一化。
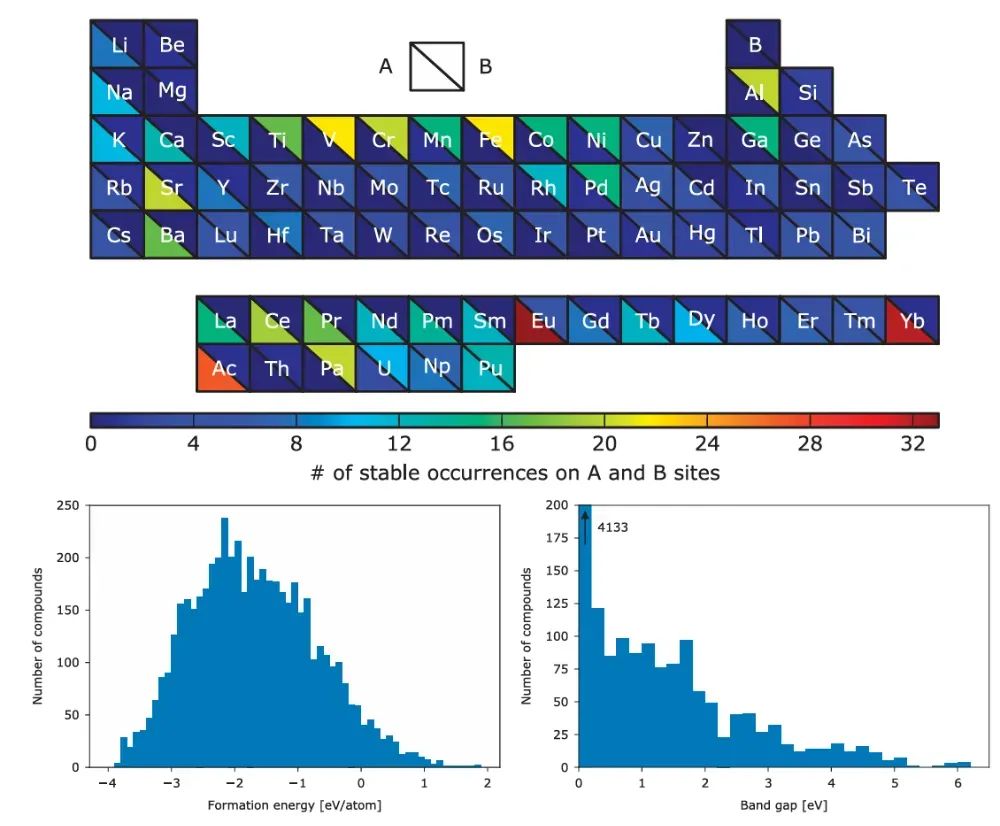
例如,Wolverton等针对~5000种ABO₃钙钛矿进行了DFT计算,得到395种热力学上稳定(凸包上的)候选材料。由此可筛选出可能实验合成的新材料。
类似地,结合能(cohesive energy)是材料和其构成原子或分子的能量差,更侧重于评价元素键合强度,但本质与形成能类似。对金属或合金,可计算不同相的总能差(如hcp与bcc金属相变的能差)来判断热力学倾向。
DOI:10.1038/sdata.2017.153
缺陷形成能:在半导体和固体材料中,点缺陷(空位、间隙、杂质等)能显著影响性能和稳定性。缺陷形成能定义为引入一个缺陷前后总能之差,通常还要考虑加/减去相应元素的化学势。缺陷形成能低表明缺陷易形成,可能引入非本征载流子导致材料性质变化。
在理论中常将缺陷形成能作图(对载流子费米能依赖)来判断哪些缺陷在特定条件下最稳定,并与实验观察(电导、光谱信号等)对应。
例如,氧化物和氮化物半导体中,氧空位或氮空位的形成能常用于解释为何某些材料自发呈现n型导电。
反应能与相变能:对多相体系或化学反应,计算两种或多种物相之间的能量差(如分解反应能、转变反应能)是评估稳定性的常用方法。若将候选相分解成其它稳定相时释放能量(反应能为负值),说明原相热力学不稳定;反之反应能为正则原相更稳定。
该思路可推广到判断同组分不同晶型之间的相对稳定性,比如Fe合金中bcc与fcc相的自由能差决定两者的稳定区域。反应能计算也可用来评估材料在不同化学潜势(如氧含量)下的稳定性区域,这被称为相图或化学势窗口分析。
DOI: 10.1103/PhysRevB.107.014102
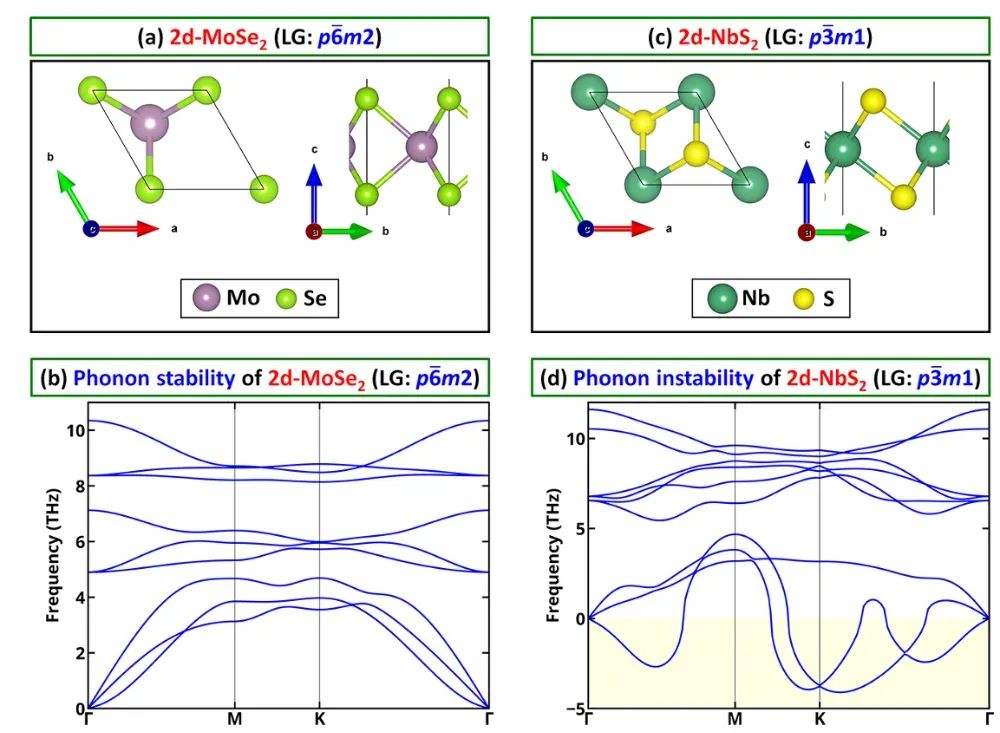
结构动力学稳定性通常通过计算声子谱来判断。声子谱(晶体的振动模式分布)在DFT框架下可由密度泛函扰动理论(DFPT)或有限差分法计算得到。
如果某结构存在虚频(声子分支出现负频率),意味着该结构对某种位移模是不稳定的,会沿虚频模式发生结构扭曲或相变。因此,无虚频是判断理论结构“动态稳定性”的普遍标准。
例如,对理论预测的新二维材料或高压相进行声子计算,常常发现若存在虚频,则该结构在室温下无法稳定存在。
声子谱还可以计算零点能和热振动自由能,在有限温度下加入对热力学稳定性的修正。由声子谱得到的热容和熵也用于更精确地评估材料在温度下的稳定性或相变温度。
AIMD模拟通过在一定温度下让体系演化来直接观察结构随热激发的响应,常用于评估材料的热稳定性。
在典型做法中,选取理想晶格结构构建超胞,在设定温度(如300–2000K)下进行NVT或NPT模拟,并监控结构是否崩塌或发生相变。
若在合理时间尺度下结构保持完整(均方位移不发散、键长无剧烈变化),则可认为该结构在该温度下热稳定;若发生分解、重组或生成液态团簇,则认为不稳定。多项研究结合声子谱和AIMD来验证预测结构的稳定性。
例如,在2D材料筛选中,Malyi等分析了一系列已知和预测的二维晶体,通过声子谱确认无虚频并通过AIMD检验室温热稳定性,从而建立了能量/声子/动力学稳定性过滤准则。
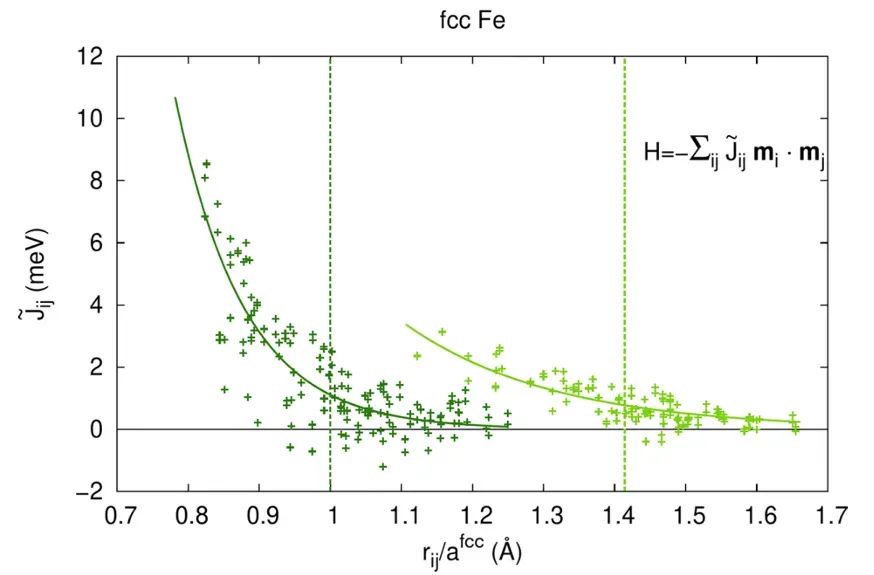
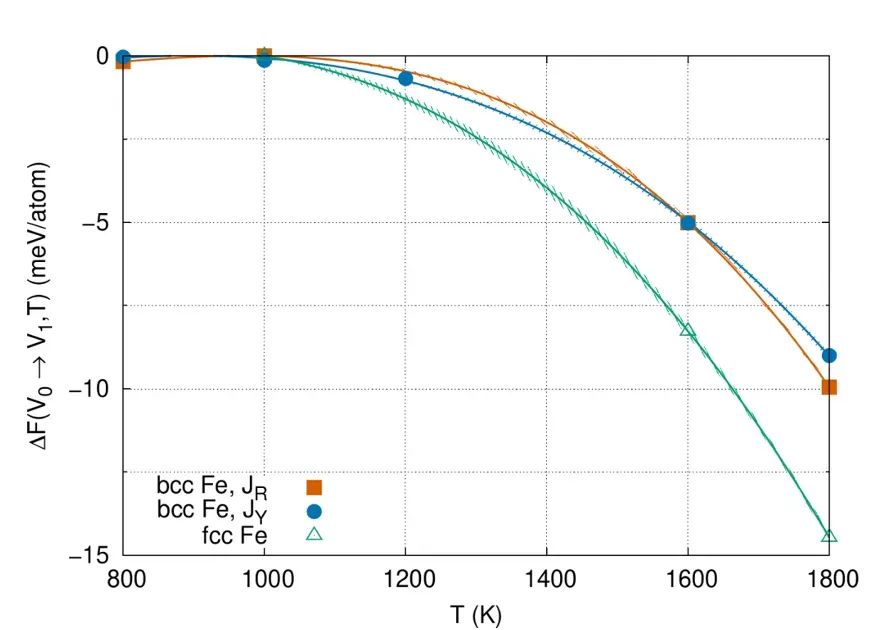
对于存在磁性或复杂电子-晶格耦合的体系,AIMD也可结合自旋动力学等方式,对相变温度进行自由能计算,如Gambino等利用AIMD结合热力学积分精确预测了Fe在800–1800K时bcc与fcc两相的稳定性。
对于纳米材料、薄膜或多相体系,表面能和界面能对稳定性起关键作用。
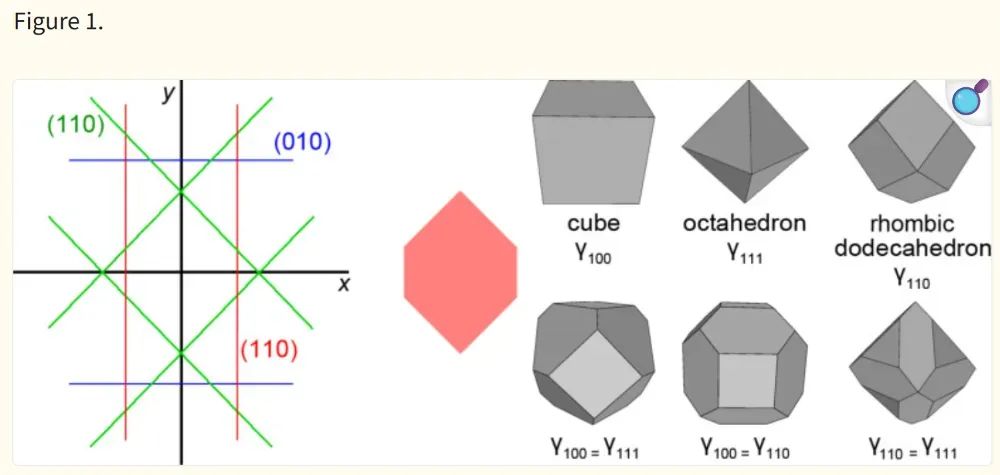
在纳米尺度时,材料的表面(或颗粒/薄膜边界)贡献大量能量,因此低表面能的晶面和稳定的界面结构会更容易形成。理论上可用DFT计算各晶面的表面能,并通过Wulff构造预测平衡晶体形貌。
Wulff构造给出一个晶体在各向同性体积约束下、使总表面能最小的形状:面能最低的晶面占面积最大。下图示意了不同面能比例下的Wulff平衡形状;例如当γ100=γ111时生成截顶八面体,当γ100≫γ111时为立方体,而当γ100≈γ111≈γ111时呈现菱形十二面体(此图不同颜色线条代表不同晶向的面能,右侧为对应的多面体形状)。
Wulff构造指出,表面能更低的晶面会在纳米粒子或晶体外形中以更大面积出现。类似地,对于异质结或多晶界面,可计算界面形成能来评估界面稳定性(如界面空位、错位等对能量的影响)。
总的来说,表面/界面能计算有助于理解纳米或薄膜材料的构型与稳定性,如某些面较易形成、某些界面易脱落等现象。
在金属材料中,DFT计算常用于评估不同晶相、合金成分与缺陷的稳定性。铁(Fe)是经典例子:在常温下体心立方(bcc)结构稳定,而高温下面心立方(fcc)稳定。
Gambino等采用自旋动力学耦合AIMD计算了两种结构在800–1800K的自由能差,精确预测出bcc→fcc相变的温度。该方法考虑了热振动和磁性自由能,为磁性金属相变提供高精度预测的范例。
另一个例子是高熵合金:理论计算可帮助筛选合金主成分,使得形成能最低,对应更稳定的均匀固溶体相。
在催化领域,研究人员还用DFT计算单原子或簇在金属表面的结合能和形成能,以评估其热稳定性和活性,比如计算Pt、Au纳米簇的面能指导形貌控制。
一般而言,金属的缺陷形成能(如空位或间隙)也用于理解扩散和相变动力学:缺陷形成能高说明金属在常温下难以自发形成高浓度缺陷,结构更稳定。
DOI: 10.1103/PhysRevB.107.014102
半导体(包括氧化物、氮化物、卤化物、硅锗等)中稳定性的评估既关注母体晶格稳定性,也关注缺陷和化学稳定性。
例如氧化物钙钛矿系列,Wolverton等对5329种ABO₃结构进行了DFT计算,结果预测出395种热力学稳定的化合物(位于能量凸包上),其中许多尚未合成。这些预测为新型光催化、电池和陶瓷材料的发现提供了指导。
对于卤化物钙钛矿(如钙钛矿太阳能材料),DFT也常用于计算形成能、离子迁移势垒等,以解释材料在温度和光照下的相稳定性。
例如,DFT表明某些掺杂或表面修饰能够抑制易形成的卤空位,从而延缓材料相分解。在传统半导体中,如碳化硅、氮化硅等,DFT可用来比较不同多晶型(3C、4H、6H等)的形成能,帮助确定哪种多晶型在特定条件下更稳。
缺陷形成能在半导体中尤为重要:它决定了载流子浓度和稳定性。比如在GaN和SiC中,理论研究发现氮空位比铝空位更易形成,说明材料往往天然呈n型;而由理论算出的高能隙绝缘体如氮化镓的某些缺陷形成能预测了实验上难以形成显著浓度的深能级缺陷。
DOI:10.1038/sdata.2017.153
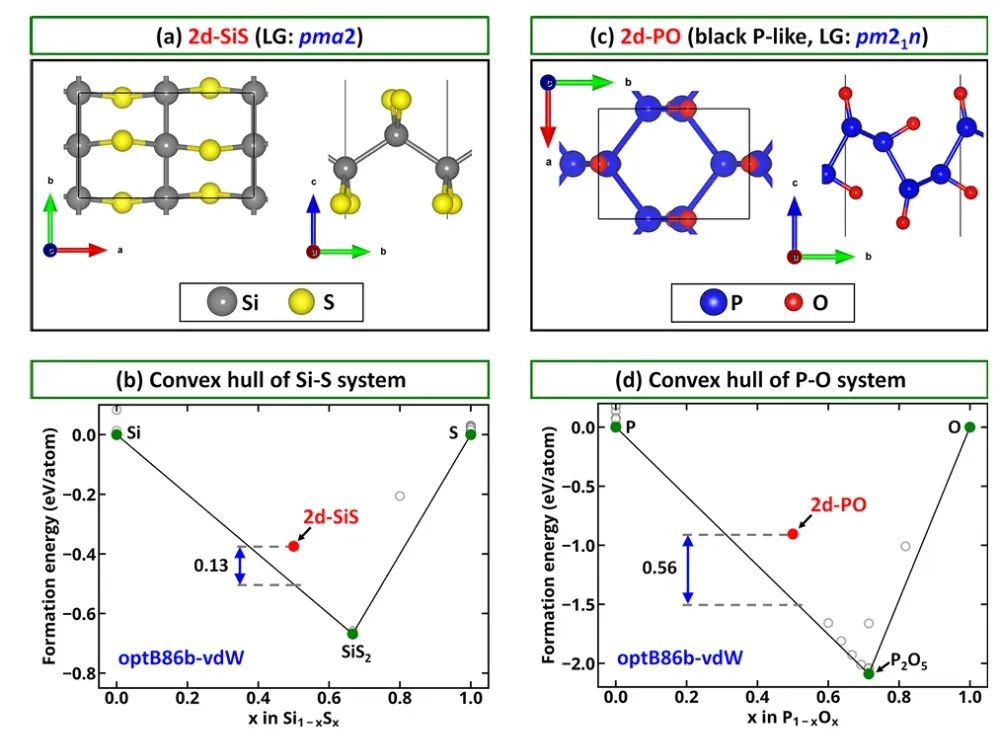
二维材料(如石墨烯、过渡金属硫化物MX₂、碳化硅烯等)具有独特的稳定性问题。由于其原子层厚度,很容易出现动态不稳定(如自由悬空单层在理论上可能发生弯曲)或化学不稳定(易氧化或水解)。
计算上,筛选2D材料时首先考察其热力学稳定性:若该二维结构可由某种3D晶体剥离得到,就计算剥离能或层间结合能,低结合能对应容易形成稳定单层。
例如,Friedrich等发现表面阳离子氧化态越低,对应层间键越弱,使得非范德华固体生成二维层时更易剥离。另一方面,声子谱计算几乎是判断2D材料稳定性的标配:若单层模型出现虚频,就意味着该层结构会发生自发畸变,难以实验制备。
因此,在现有2D数据库或计算筛选中,只有通过声子谱测试(确保无虚频)的材料才被认为“初步稳定”。
DOI:
10.1021/acs.nanolett.1c03841
理论计算不仅用于预测新材料,更重要的是解释和反推实验结果。当实验中观测到相变或结构演化时,可以使用DFT和相关计算来阐明其原因。
例如,若高温实验出现某种新相,计算不同相的能量可判断哪个相在该条件下更稳定;若快速淬火后得到的相与平衡相不同,可计算相变动能垒和自由能差来解释该相为何能够“滞留”成为过冷相。
在实际案例中,如果实验发现某异质结在特定化学环境下分解,可以通过DFT计算界面反应能或元素扩散路径来再现这一过程。
总之,通过对计算结果(能量、力学、电子结构等)的分析,我们可以为实验中观察到的材料相变、形貌演变或稳定性差异提供合理解释,进而指导实验策略。
理论计算与实验观测通常是互补的:理论能预测可能的稳定相、筛选材料,而实验验证所筛选结构是否真实存在。
在实验方面,理论结果可以解释复杂现象:如为什么某一非晶薄膜在退火时转变为特定晶相(因为DFT计算显示该晶相自由能最低),或为什么腐蚀性试剂会引起特殊表面结构变化(因为DFT揭示某表面结构在该条件下形成能最低)。
哪些现象适合引入理论协助?一般而言,当实验遇到如下情况时,理论辅助分析特别有效:①结构判定:新相或新结构的性质预测与验证;②相变机理:热力学与动力学因素共同作用下的相态图;③缺陷与掺杂:预测缺陷种类与浓度对性能的影响;④表面与界面现象:催化活性、纳米形貌、异质结构的热力学稳定性等。
在这些情景下,使用DFT或高级模拟能提供微观机理和能量分析,为实验提供量化依据。理论-实验协同已成为现代材料研究的重要趋势:高性能期刊如Nat Mater、PRL、Nano Lett中不乏实验论文配合理论计算解释的案例,强调了二者结合的重要性。
总之,理论计算为实验研究者提供了强大的工具,不仅可以筛选和预测稳定性高的材料候选,还能解释实验观测到的稳定性差异或相变过程,从而加速材料发现并深入理解材料行为。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!