



单原子催化剂(SACs)具有原子分散的活性位点,能够高效地将小分子转化为高附加值的燃料和化学品。单原子位点(SASs)的催化性能从根本上受金属中心的配位几何结构与催化中间体吉布斯自由能之间的协同作用所支配。
SASs的固有活性不仅取决于中心金属物种,还依赖于包括第一、第二和更高配位壳层在内的层次化配位环境。具体来说,第一配位壳层通过金属中心的p-d轨道杂化,使得电荷重新分布以精确调节d带中心位置;第二配位壳层则通过部分电荷转移至第一配位壳层中的异原子间接影响催化行为,从而在活性金属位点引发二次电子扰动。
尽管大量研究致力于构建这些近邻配位壳层,但更高阶配位壳层(>第二配位壳层)在介导电子性质和反应能量方面的作用仍很少探讨。
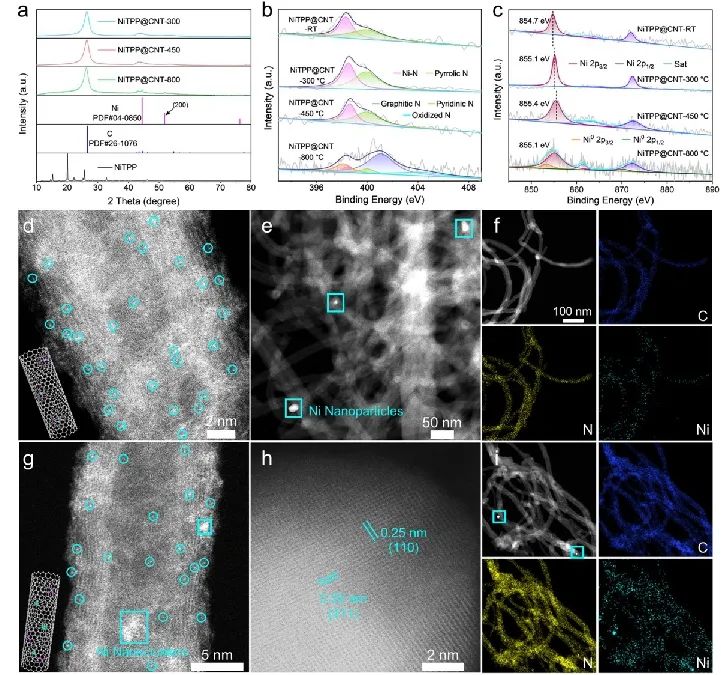
近日,中国科学技术大学吴宇恩、深圳大学余振强和国防科技大学黄浩等使用具有明确Ni-N4构型的四苯基卟啉镍(NiTPP)作为前驱体,报道了一种合理的配位工程策略。通过温度依赖的热解研究,研究人员确定了一个选择性裂解苯基与β-C位点之间C-C单键的分解机制。
这种可控的裂解保留了主要的Ni-N配位(第一壳层)和次要的C配位(第二壳层),同时系统地消除了较高配位壳层中的π电子网络。这种精确定制的电子环境使得催化剂具有卓越的CO2电还原性能,在-1.0 VRHE下,CO选择性达到99.4%。
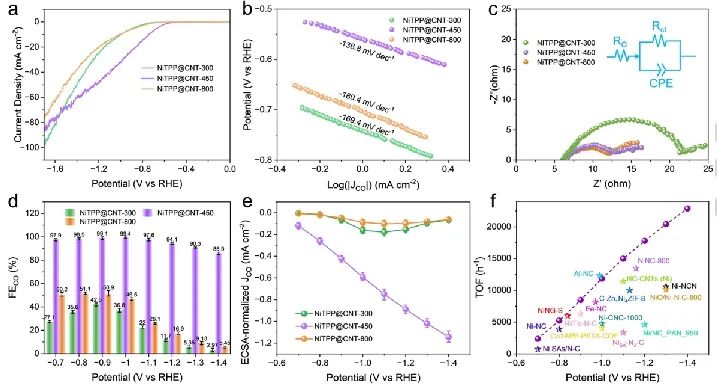
值得注意的是,在增强的反应条件下(-1.4 VRHE),最优的催化剂的CO法拉第效率提高了29倍(从2.97%增加到85.9%),同时周转频率增加了18倍(从1248 h-1增加到22881 h-1),这证明了Ni活性位点内在活性的提升。
此外,制备的催化剂在工业相关条件下同样表现出优异的性能,在流动池中和500 mA cm-2电流密度下维持98.3%的CO选择性。原位红外光谱测量显示,去除外围π电子后,*COOH中间体加速生成以及促进CO在Ni位点的解吸。
密度泛函理论(DFT)计算进一步表明,长程π电子离域的消失有效增强了CO2的吸附,同时降低了*COOH中间体形成的能垒,加速反应动力学。
Beyond second coordination shell: Long-range π-electrons delocalization engineering in single-atom catalysts for CO2 electroreduction. Angewandte Chemie International Edition, 2025. DOI: 10.1002/anie.202506663