本文阐述了使用GGA-PBE泛函进行DFT带隙计算时常出现比实验值低约40%~50%的系统性偏差,其根本原因在于常规模型缺乏“导数不连续”校正且存在自相互作用误差。
随后指出尽管绝对带隙值偏低,但由于这种低估对不同材料和结构变化均保持一致性,PBE计算依然能够可靠地反映带隙随掺杂、应力或维度变化的相对趋势(如2D SnTe层数减小带隙增大的现象)。
文中通过界面模型和态密度分析进一步说明了PBE在能带色散和轨道贡献解析方面的价值。
最后简要介绍了诸如杂化泛函(HSE06)、GW方法、DFT+U、meta-GGA和mBJ势等多种可提升带隙预测精度的替代方案,并强调在材料研究中可根据需求,将PBE用于快速筛选与定性趋势判断,高精度方法用于关键定量预测。
在材料科学领域,理论计算是一种非常重要的研究手段,尤其在探索新材料和分析实验现象时具有不可替代的作用。
然而,许多实验背景的研究人员发现,当使用广义梯度近似的Perdew–Burke–Ernzerhof泛函(GGA-PBE)进行能带计算时,往往得到的禁带宽度(带隙)比实验观测到的数值偏低。这种差异常引起误解,甚至质疑计算结果的可靠性。
实际上,这并非计算本身的错误,而是由计算方法的内在局限性导致的。下面将详细解释这一现象的原因,并探讨尽管存在这一系统偏差,GGA-PBE方法仍然具有的重要应用意义,同时简要介绍几种更准确的计算方法供研究者参考。
使用广义梯度近似的PBE功能(GGA-PBE)进行密度泛函理论(DFT)计算时,常常会得到比实验值偏小的带隙(禁带宽度)。这并非计算错误,而是DFT近似本身的局限所致。
首先,DFT是一种基态理论,旨在准确预测体系的最低能量状态。带隙则涉及将电子从价带激发到导带的激发态性质,严格来说不在标准DFT直接优化的范畴。
常用的局域或半局域交换-相关泛函(如LDA和GGA)无法精确描述增加或移除一个电子时体系能量的突变,这意味着缺少所谓的“导数不连续”项,导致Kohn-Sham能级计算出的带隙低于真实的光学带隙。
此外,GGA-PBE中还存在自相互作用误差:电子与自身产生的库仑势未被完全抵消,使价带电子在计算中被过度“抹平”或离域,能量高于应有值,从而使价带顶与导带底的能量差减小。
简而言之,PBE这种近似方法倾向于低估电子间的排斥作用,导致计算得到的价带和导带能级距离(即带隙)比实际情况更近。
这一现象在半导体和绝缘体中相当普遍,是DFT的知名问题之一。
用PBE计算常见半导体的带隙往往比实验值低40%~50%。晶体数据库Materials Project的内部测试显示,PBE预测的带隙平均比实验低约40%,有些已知的窄带隙绝缘体甚至被预测成金属态。导致低估的主要原因正是上述泛函近似和方法学上的局限。
因此,当使用GGA-PBE计算材料带隙时,需要认识到这属于方法本身的系统性偏差,而不是具体材料或计算步骤的问题。
尽管PBE给出的绝对带隙偏低,但这些计算结果在材料研究中仍有重要的参考价值。
首先,这种低估通常具有一致的系统性,也就是说对不同结构或成分的材料都会类似程度地低估。
因此,我们可以比较不同条件下带隙的相对变化:例如,研究掺杂、应力或结构缺陷对带隙的影响时,PBE计算虽然绝对值不准,但可靠地反映出哪种情况带隙更大或更小。
这样的趋势比较对于材料改性很有意义——如果计算显示某种掺杂使带隙明显变宽,我们可以相信这种趋势,即使具体数值有所偏差。
在实际研究中也确实如此,有实验发现材料经过掺杂后带隙变宽或变窄的方向与PBE计算预测的趋势一致。
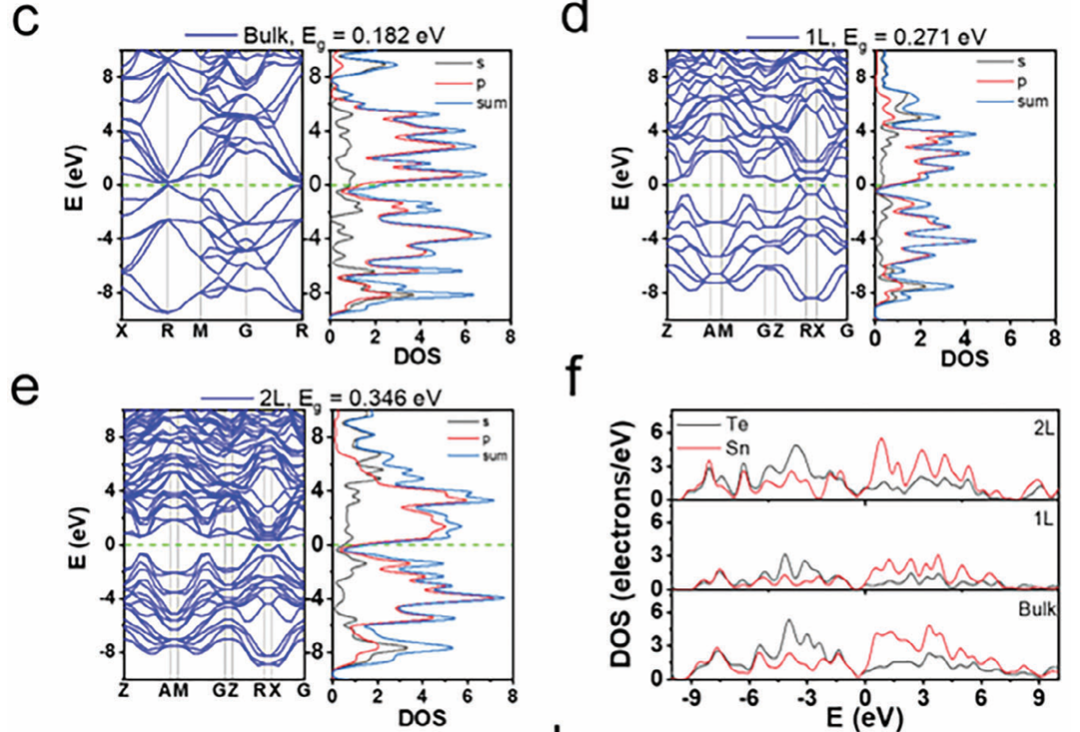
Singh 等人(2024)针对二维锡碲(2D SnTe)材料的热电性能开展了系统研究,进一步验证了 GGA-PBE 在带隙预测中虽存在绝对值偏低的缺陷,但在结构演化趋势判断上具有高度可靠性。
他们通过计算不同层数(块体、单层和双层)SnTe 的能带结构,发现 GGA-PBE 预测的块体 SnTe 带隙仅为 0.18 eV,而该材料的实验带隙普遍约为 0.6 eV,显示出明显低估。
然而,随着层数减小,PBE 计算带隙有序增加至 0.27 eV 和 0.35 eV,与实验观察的“维度减小时带隙变宽”的趋势完全一致。
此外,研究还指出,二维量子限域引起的电子结构变化显著增强了 Seebeck 系数,而这一增强趋势也被 PBE 成功捕捉。
这进一步表明,尽管 PBE 低估了带隙绝对值,但其在结构调控(如厚度变化)导致的电子性能趋势分析中,仍具有良好的系统性与参考价值。
DOI: 10.1002/smll.202403728
其次,电子结构的其他方面信息如能带排列、能带形状(色散关系)和态密度分布等,PBE计算通常能够较准确地给出。这些性质往往主要由价带和导带的能级顺序及成分决定,受带隙绝对值影响较小。
实际上,DFT计算的能带色散(能带随$k$的变化)在很多情况下与实验观测符合得很好,能量误差一般在0.1~0.4 eV范围。因此,我们可以从PBE计算的能带图中判断材料是直接带隙还是间接带隙半导体、哪种轨道组成了导带底/价带顶,以及能带宽度等趋势。
这些定性或半定量信息对理解材料的导电性、光学跃迁等性质非常有用。态密度(DOS)曲线也能显示价带和导带各能级的分布情况,帮助识别如缺陷能级、费米能级位置等。
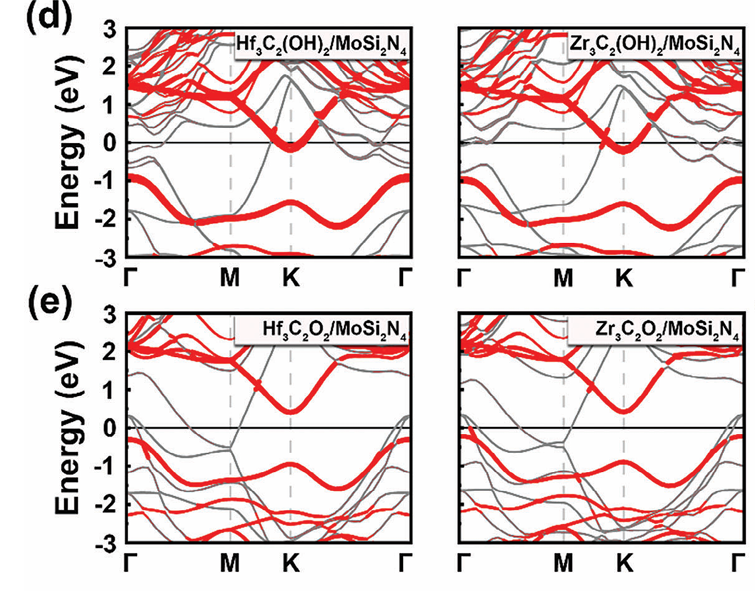
Shan等人于2024年发表在 Advanced Functional Materials 的研究中,系统地构建了二维金属(Mxene)与二维半导体XSi₂N₄之间的界面模型,并利用GGA-PBE方法对其能带结构、态密度(DOS)、界面电荷转移等电子特性进行了深入分析。
尽管PBE低估了带隙,但其能带图中的导带底(CBM)和价带顶(VBM)依然准确地反映了由Mo–N内层贡献主导的态密度分布,而外层Si–N亚层起到了“电子缓冲保护”的作用,使得界面电子态不易受到金属接触面态的扰动。
这不仅确保了带边态的有序性,也使得其态密度图中能清晰地区分材料本征态与杂化态的能级分布。
文中还进一步表明,即便在构建共价键型接触后,PBE预测的能带结构变化趋势依旧与实验或高精度方法(如COHP分析)所得结果一致,强化了PBE计算在“相对比较”和“轨道解析”方面的可信度。
DOI: 10.1002/adfm.202412773
在需要与实验带隙对比时,研究者有时会应用一个简单的“剪刀操作”(人为将计算的导带整体上移,使带隙与实验值对齐),以在保持PBE预测的能带形状不变的前提下校正带隙,用于进一步的光学谱计算等。
这进一步体现了PBE计算提供的基准电子结构具有价值——哪怕带隙本身偏低,我们依然可以利用其结果进行各种比较和分析,从中获取材料的趋势性结论和物理图像。
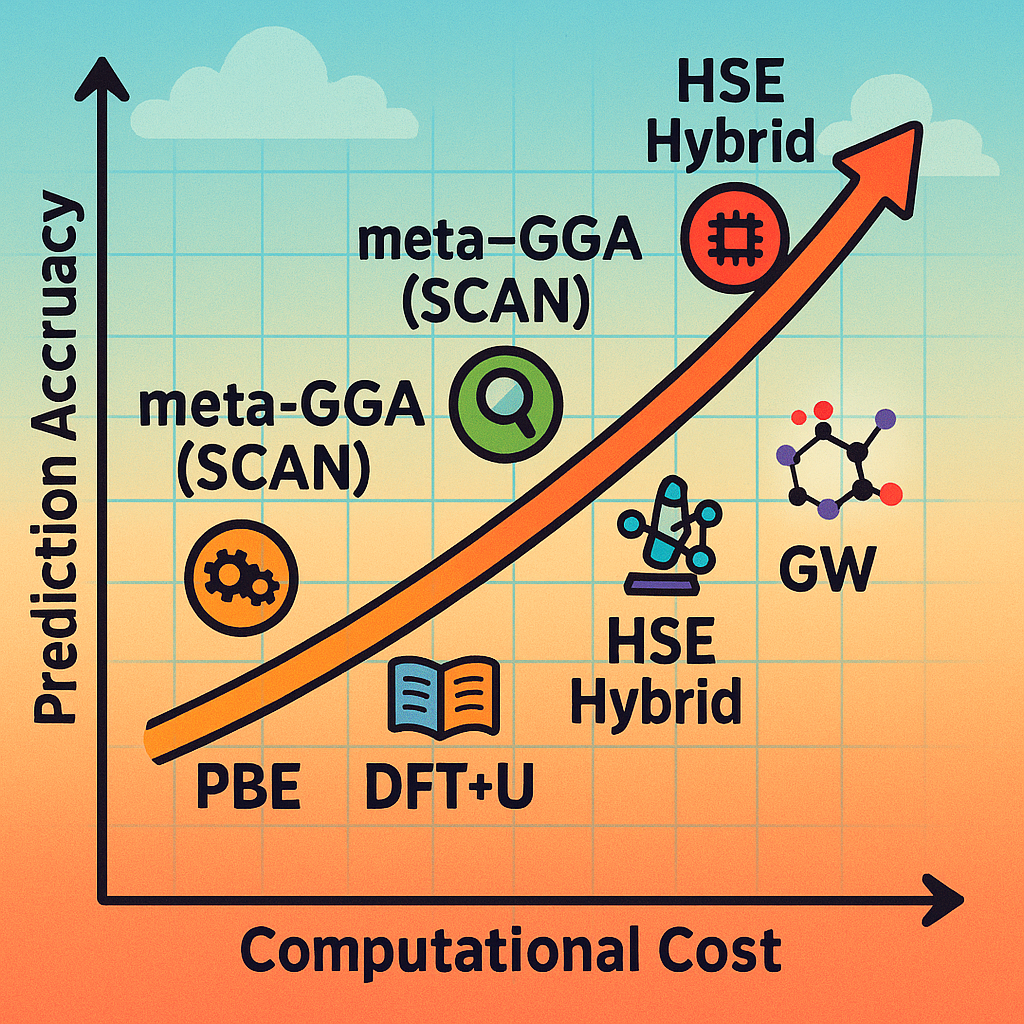
针对DFT-PBE带隙低估的问题,已经发展出多种更准确的计算方法来获得更接近实验的带隙值。以下简要介绍几种常用的方法及其特点:
杂化DFT泛函(如HSE06):杂化泛函通过将一部分精确交换(Hartree-Fock方法的交换项)混入常规DFT的泛函中来提高精度。
直观地说,纯DFT(如PBE)的交换-相关作用偏弱导致带隙偏小,而纯Hartree-Fock方法又因忽略电子相关作用往往高估带隙;杂化方法适当混合两者可以在二者之间取得平衡。
例如,PBE0泛函采用25%的Hartree-Fock交换,HSE06则在此基础上进一步引入截断(range-separation),仅对电子相互作用的短程部分采用精确交换,从而在保持精度的同时降低计算量。引入精确交换可以减轻自相互作用误差,显著改善计算的带隙并给出更合理的能级排列。
杂化泛函通常能将带隙预测提升到接近实验值的水平(相对于PBE有几十%的提高),但计算资源的消耗也比GGA大数倍。
GW准粒子方法:GW是一种多体微扰论的方法,用于计算固体的准粒子能量。名称中的G和W分别代表格林函数和屏蔽相互作用。
简单来说,GW方法考虑了电子-电子间的动力学相互作用和屏蔽效应,通过计算电子的自能校正来调整Kohn-Sham能级,使之更接近真实的电子激发能量。
由于GW能更准确地描述一个电子加入或移出体系所需的能量,它通常预测出比PBE更大的带隙,更贴近实验值。GW被认为能够部分弥补DFT中缺少的导数不连续,为带隙计算提供高精度的准粒子描述。
然而,其代价是计算非常耗时,需要远多于DFT的计算资源,因此常在研究中作为后续校正步骤(例如PBE计算完电子结构后,再做单点GW校正)使用。
其他改进方法:除了以上两类主流方法外,还有一些针对特定情况的修正手段。
例如DFT+U方法在常规DFT哈密顿中加入一个Hubbard U项,用以加强对局域d或f电子的束缚。这样可使过于离域的价电子重新局域,降低其能量,从而扩大间隙,一定程度上修正LDA/GGA带隙低估的问题。
DFT+U方法对过渡金属氧化物等强关联体系尤其有效,计算开销与普通DFT相当,但需要事先选定合适的U值。
另一些新的密度泛函如meta-GGA(元泛函)和半经验的mBJ势(改进Becke-Johnson势)也被证明可以给出比PBE更准确的带隙,它们通过改进泛函形式或引入经验参数来部分恢复导数不连续效应。
这些方法各有优缺点:精度上通常介于PBE和GW之间,而计算成本介于杂化泛函和GW之间。研究者可以根据需求选择合适的方法,在追求精度和控制计算资源之间取得平衡。
综上,尽管GGA-PBE低估了材料的禁带宽度,但我们理解了这是理论近似的固有现象,并可以善加利用PBE的计算结果进行趋势分析和机制研究。在需要更准确数值时,可考虑采用杂化泛函、GW等更高精度的方法。
对于主要从事实验的材料科学研究者而言,这样的计算结果依然提供了丰富的信息和指导意义:PBE提供基准的电子结构图景,而更先进的方法则为精确预测提供了手段。
通过理论与实验的合作,我们既能用PBE快速筛选和比较材料,又能在关键处借助高精度计算获得与实验吻合的定量结果,从而更全面地理解材料的电子结构特性。
课程基于CASTEP, Dmol3模块设计,专门讲解正、负极材料计算研究(另一部分为电解液/固态电解质的设计)。课程内容包括电压曲线、克容量、离子迁移、过渡态、复合材料、姜泰勒效应、阴阳离子氧化还原、离子吸附分析、磁性等内容,重现案例来自PRB、Advanced Materials等期刊。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!