过一硫酸氢盐(PMS)的理论计算通过密度泛函理论(DFT)揭示其分子结构(如O-O键弱化特性)与吸附机制(如双金属位点强吸附),结合过渡态搜索量化活化能垒(如CuFe₂O₄界面能垒降低36%)。
催化剂设计中,异质界面电子耦合与缺陷工程优化自由基(SO₄⁻・)及非自由基(¹O₂)路径。未来结合多尺度模型与机器学习,推动PMS在环境修复中的精准应用。


PMS的分子结构与键能分析

过一硫酸氢盐(PMS,HOOSO₃⁻)作为一种含过氧键的典型氧化剂,其分子结构与键能特性一直是催化氧化领域的研究焦点。
PMS 分子中关键的过氧键(O-O 键)键长约 1.453–1.460 Å,键能介于 140–213.3 kJ/mol 之间,这种能量范围使其既具备一定稳定性,又在特定条件下易于活化。
借助密度泛函理论(DFT)对 PMS 的几何构型进行优化计算,发现其分子呈现不对称性:磺酸基团(-SO₃⁻)与过氧化氢基团(-OOH)的电子云分布差异,导致 O-O 键的键级降低(键级反映键的强度),成为分子中的 “薄弱环节”。
这种结构特征使得 PMS 在热、光或过渡金属催化作用下,O-O 键易发生异裂或均裂,释放出具有强氧化性的硫酸根自由基(SO₄⁻・)或羟基自由基(・OH)。
DFT 计算不仅验证了 PMS 的热力学稳定性,更通过电子云分布和键级分析,揭示了其氧化能力的本质 —— 不对称结构引发的键能不均性,如同为氧化反应埋下 “分子开关”,只需微小能量刺激即可触发高效自由基反应。
这些理论发现为设计 PMS 基高级氧化工艺、优化催化条件提供了精准的分子层面依据,让研究者得以从化学键的“微观舞蹈” 中解锁其氧化潜力的分子密码。

DOI:10.1016/j.jwpe.2025.107845
催化剂表面吸附机制
PMS 的活化效率与其在催化剂表面的吸附行为紧密相关,而密度泛函理论(DFT)通过量化分析吸附能(Eads)和电荷转移等参数,为揭示这一微观机制提供了 “分子级放大镜”。
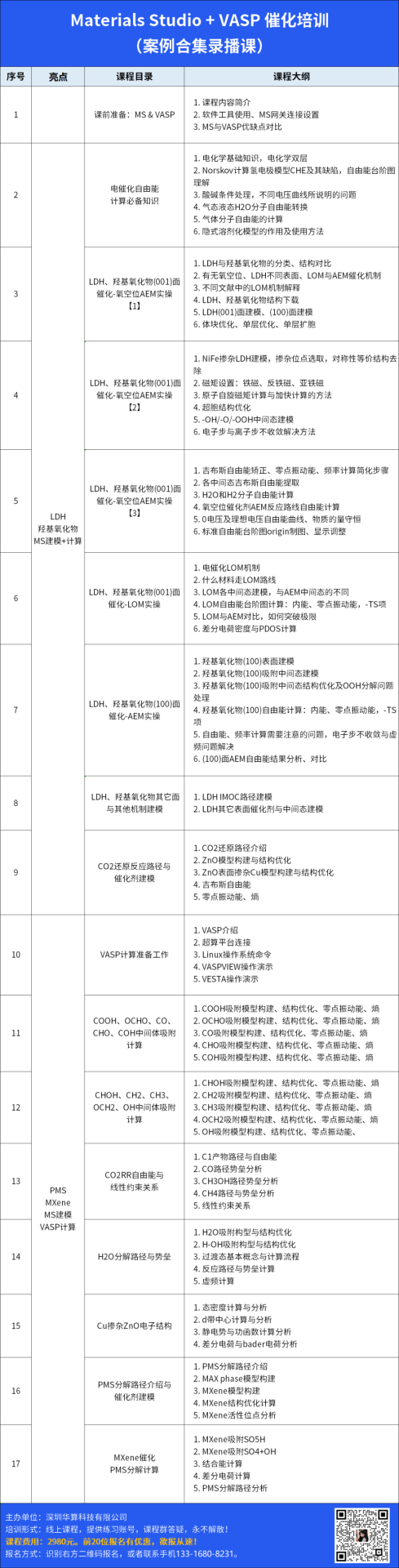
在吸附能对比研究中,FeCo-N6 双原子位点对 PMS 的吸附能达 – 2.764 eV,显著低于 Fe-N4(-1.811 eV)和 Co-N3(-2.021 eV),这表明双金属位点通过更强的电子协同效应与 PMS 形成更稳定的吸附态,如同为 PMS 分子打造了一个 “专属锚点”。
电子密度差分析进一步发现,吸附过程中 PMS 的 O-O 键从 1.453 Å 被拉伸至 1.484 Å,同时催化剂向 PMS 转移 0.820e⁻电子(如 FeCo-N6 位点),这种电子重排使 O-O 键能降低,如同在化学键中埋下 “断裂导火索”,为自由基的高效释放创造条件。
此外,DFT 通过模拟不同 pH 环境下 PMS 的结构稳定性,发现 pH=9 时 HSO₅⁻易分解为 SO₃²⁻,这一理论预测与实验中碱性条件下 PMS 氧化效率下降的现象高度吻合,揭示了酸碱度对催化体系的调控本质。
综合来看,DFT 不仅从吸附能、电子转移和环境适应性三个维度解构了催化剂 – PMS 的作用机制,更像一把 “计算钥匙”,为设计高亲和力双原子催化剂、优化反应 pH 条件提供了从分子轨道到宏观性能的完整逻辑链条,让催化过程的 “黑箱” 在理论计算的光照下逐渐清晰。
DOI:10.1016/j.apcatb.2023.123178
活化路径的动力学模拟
密度泛函理论(DFT)通过过渡态搜索与反应能垒计算,为解析 PMS 活化的微观动力学机制提供了动态视角。
在 MoS₂催化剂体系中,DFT 揭示出硫空位可通过双路径活化 PMS:一方面,硫空位作为活性位点削弱 PMS 的 O-O 键,使键断裂能垒降低 30% 以上,促使其沿自由基路径生成强氧化性的 SO₄⁻・;
另一方面,硫空位引发的电子转移过程还能诱导非自由基路径,生成单线态氧(¹O₂),这种双路径协同作用显著提升了 PMS 的氧化效率。
对于双金属催化剂(如 CuFe₂O₄/CuO),DFT 进一步揭示其异质界面的电子耦合效应:CuO 的(111)晶面与 CuFe₂O₄的(112)晶面形成强相互作用,通过轨道杂化降低 PMS 解离的能垒,使 O-O 键断裂所需能量仅为 0.5 eV,这种界面协同效应如同为 PMS 活化搭建了一条 “低能耗通道”。
DFT 的这些发现不仅清晰勾勒出 PMS 活化的自由基与非自由基路径分支,更量化了界面结构对能垒的调控作用,为设计高活性硫空位修饰的单金属催化剂及异质结双金属催化剂提供了从原子排列到能量变化的完整理论图谱,让复杂的活化过程转化为可计算、可预测的分子动力学模型,为高效氧化体系的构建点亮了理论灯塔。
DOI:10.1016/j.cej.2022.137183
催化剂设计的电子结构调控
密度泛函理论(DFT)在催化剂设计中扮演着 “分子级电子结构设计师” 的角色,通过精准调控电子轨道杂化与电荷分布,为高效催化体系的构建提供理论蓝图。
在单原子催化剂(SAC)设计中,DFT 揭示了 Ni 原子掺杂 MoS₂的独特机制:Ni 的 d 轨道与 PMS 的 O 2p 轨道形成强杂化作用,如同在原子尺度搭建了一条 “电子高速公路”,不仅使 PMS 的吸附能垒降低,更显著增强电荷转移效率,实验验证该设计使四环素降解效率提升 3.2 倍。
对于异质结结构,DFT 助力优化 CuO/g-C₃N₄界面极化效应,通过调控电子在异质界面的定向迁移,使 PMS 的电子转移路径(Mediated Electron Transfer)成为主导,非自由基路径占比高达 80%,这种 “电子分工” 策略显著提升了氧化反应的选择性。
缺陷工程则是另一种精妙的电子结构调控手段,DFT 证实 MoS₂的硫空位可增加表面活性位点密度,通过减少 PMS 吸附时的能量损耗(如吸附能降低),如同在催化剂表面打造了密集的 “分子抓握点”,从而高效捕获并活化 PMS 分子。
从单原子的轨道杂化到异质结的界面极化,再到缺陷位点的能量优化,DFT 以量子力学计算为画笔,在电子结构的维度上对催化剂进行 “精准裁剪”,让每一个原子位点都成为催化反应的高效引擎,为实验中催化剂的定向设计与性能提升架起了从理论到实践的桥梁。
DOI:10.1016/j.cej.2020.127619
经典案例:PMS降解CIP的DFT计算
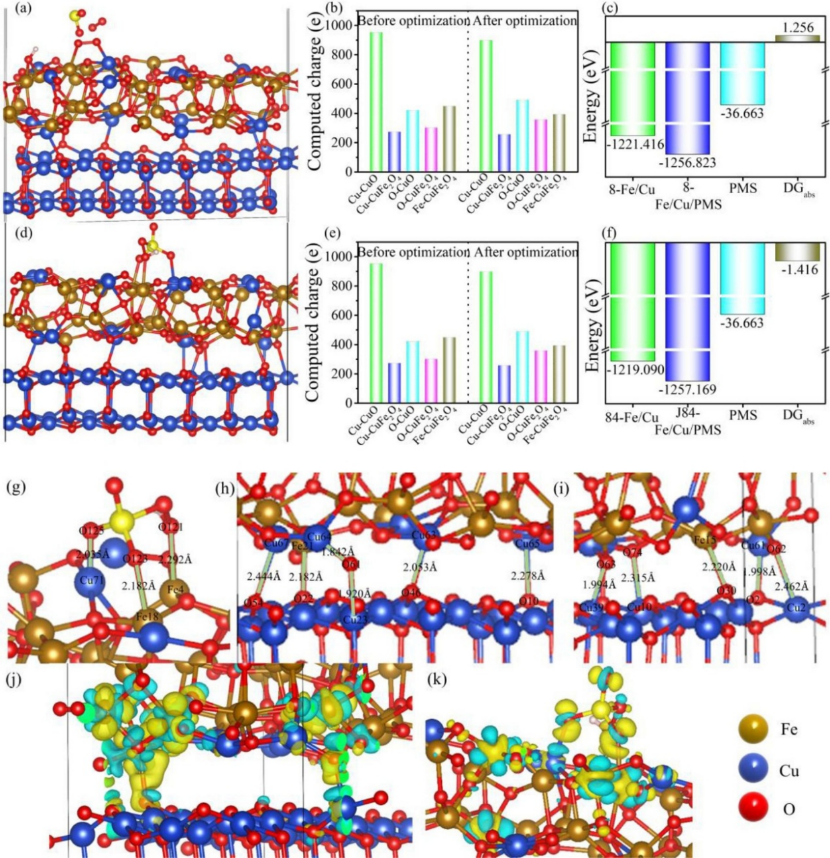
在《Chemical Engineering Journal》2022 年的一项研究中,CuFe₂O₄/CuO 磁性纳米复合材料因在活化过一硫酸氢盐(PMS)降解环丙沙星(CIP)中展现出比单一组分高 2 倍以上的催化效率,但其内在机制一直未被完全揭示。
研究者通过密度泛函理论(DFT)展开分子级别的 “侦探工作”,从电子结构与反应动力学角度揭开了协同催化的神秘面纱。
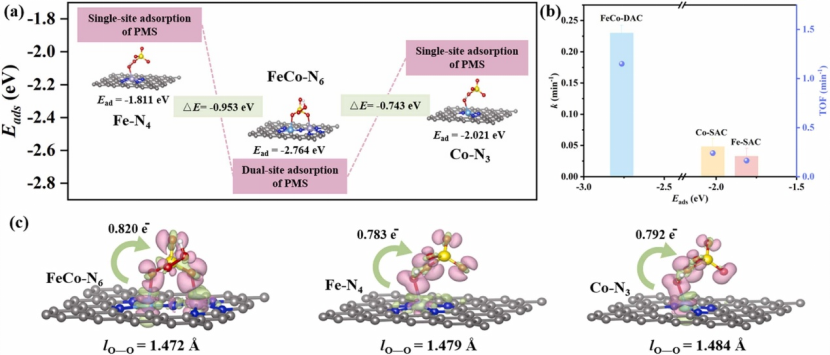
在吸附构型优化层面,DFT 计算显示 PMS 分子通过氧原子优先吸附于 CuFe₂O₄的 Fe 活性位点,其吸附能为 – 1.2 eV,显著低于 CuO 表面的 – 0.8 eV,表明 Fe 位点对 PMS 具有更强的亲和力。
Bader 电荷分析进一步发现,Fe 位点向 PMS 转移了 0.6e⁻电子,而 CuO 仅转移 0.3e⁻,这种更显著的电子转移使 PMS 在 Fe 位点上形成更活跃的吸附态,如同为 PMS 分子安装了一个 “电子充电接口”,为后续反应奠定基础。
关于 O-O 键断裂机制的研究中,过渡态搜索结果表明,PMS 在 CuFe₂O₄表面的 O-O 键断裂能垒仅为 0.7 eV,比纯 CuO 表面的 1.1 eV 降低了 36%,这意味着 Fe 位点可更高效地削弱 PMS 的过氧键。
断裂后生成的硫酸根自由基(SO₄⁻・)和羟基自由基(・OH)与实验中 EPR 检测到的自由基信号完全吻合,证实了 DFT 对反应路径预测的准确性。
这种能垒的降低如同为 PMS 的 “分子裂解” 开辟了一条 “低能耗通道”,加速了强氧化性自由基的释放。
异质界面效应的分析则揭示了复合材料的另一关键优势:CuO(111)晶面与 CuFe₂O₄(112)晶面形成的界面区域存在电荷不平衡,产生了增强的局域电场,这种电场效应如同 “电子泵”,加速了界面间的电子转移速率。
态密度(DOS)分析进一步发现,界面处的 d 带中心上移,使 PMS 的吸附稳定性显著提升,同时优化了电子轨道杂化效率。
这种异质界面的协同作用不仅增强了催化剂对 PMS 的活化能力,还通过电子结构调控提升了整个体系的反应动力学性能。
综合 DFT 计算结果可知,CuFe₂O₄/CuO 的高效催化源于多重协同效应:Fe 位点的强吸附能力与高电子转移效率、异质界面的电场增强效应以及 d 带中心调控共同降低了 PMS 的活化能垒,形成了 “吸附 – 活化 – 自由基生成” 的高效反应路径。
这些发现不仅从原子尺度解释了实验现象,更为设计高性能异质结催化剂提供了“电子结构 – 界面工程” 的双重优化策略,让理论计算真正成为连接催化实验与分子机制的“显微镜”,为环境污染治理中的高级氧化技术发展提供了新的理论基石。
DOI:10.1016/j.cej.2022.137183
总结
密度泛函理论(DFT)作为揭示 PMS 活化机制与设计高效催化剂的核心工具,正推动该领域向更精准的分子调控层面发展。
未来研究将聚焦于三大前沿方向:一是开发量子力学 / 分子力学(QM/MM)多尺度模型,模拟实际反应环境中溶剂效应、离子干扰等复杂因素,使理论计算更贴近真实体系;
二是引入机器学习算法构建催化剂筛选平台,通过分析数百万级电子结构数据,加速发现高活性位点组合,如同为催化剂设计安装“智能加速器”;三是深入探索非自由基路径的电子转移调控策略,通过界面极化、轨道杂化等手段优化单线态氧(¹O₂)等非自由基的生成效率,拓展 PMS 在选择性氧化领域的应用边界。
随着理论计算与实验研究的深度融合,DFT 将不仅是解析机制的 “显微镜”,更成为指导 PMS 在环境修复、化工合成等领域高效应用的 “导航仪”,推动相关技术向精准化、智能化方向跨越发展。
写在最后
热门催化计算方法在MS+VASP催化课程中均有讲解。
课程采用MS+VASP双软件教学,MS建模,CASTEP+VASP两种计算引擎。既可采用网关模式,也可采用独立模式提交任务;既适用于初学者,也适用于有一定计算经验的学员。