首先阐述了DFT计算吸附能、反应自由能及活化能等指标如何揭示反应热力学与动力学特征。接着基于描述符分析与火山图理论,说明了高通量DFT筛选策略在快速锁定最优催化剂候选中的应用。
随后概述了典型的DFT–实验协同工作流程及其在电化学催化(如ORR、HER、CO₂RR)和热/光催化领域的成功案例。最后展望了DFT与数据驱动方法(如机器学习)结合的发展前景,为催化剂设计提供高效、定量化的理论指导。
催化剂的设计和筛选对能源化学与环境化学等领域至关重要。然而传统的“试错”式实验方法耗时费力。密度泛函理论(DFT)作为一种高效的量子化学计算工具,可以从原子和电子层面模拟催化过程,为催化剂设计提供指导。
DFT 能够计算催化剂材料及吸附物的总能量,从而得到反应自由能、活化能、吸附能等关键指标。这些计算结果帮助研究者理解反应热力学和动力学,预测催化剂性能,并在实验验证前对大量候选材料进行预筛选。
吸附能(Adsorption Energy):指反应中间体或反应物分子吸附在催化剂表面时的结合能。
根据萨巴蒂埃原理(Sabatier 原理),理想催化剂对中间体的结合应适中:结合过弱无法有效活化反应,结合过强则不利于反应产物的解吸和进一步转化。DFT 可直接通过计算吸附体系总能量与气相能量之差来得到吸附能:
Eads=Ecat+ads–Ecat–Eadsorbate
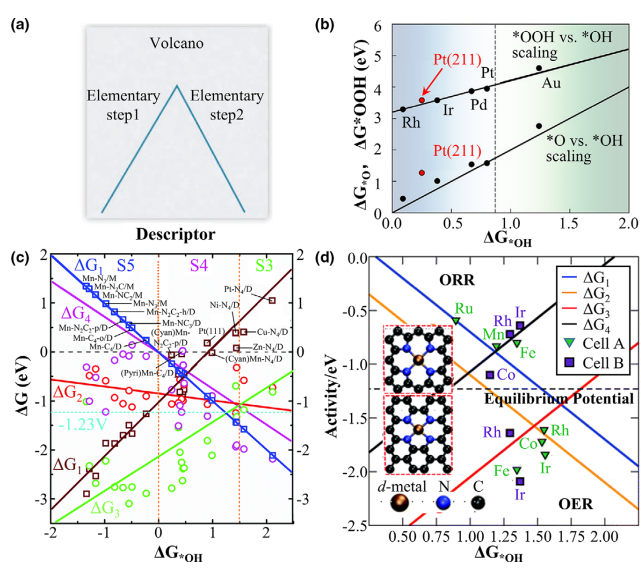
对应吸附自由能 ∆Gads,负值表示吸附是放热过程。吸附能常常用作催化活性描述符,例如在氧还原反应(ORR)中,*OH 的吸附自由能经常作为评价活性的指标,其最优值对应于火山图峰顶位置。
DFT 计算还常发现不同吸附中间体之间的线性标度关系(如 *OOH、*OH、*O 吸附能线性相关),从而简化了描述符的数量。
反应热力学自由能(Reaction Free Energy):指催化反应各步从反应物到产物的自由能差(如 ∆Greaction}。它决定反应是否自发和最终平衡:∆G
通过DFT计算初态、末态能量,可评估整个反应序列的热力学可行性。某些反应需要通过电化学电位才能“驱动”进行,此时DFT得到的自由能变化直接对应需要的过电位。
例如,在CO2还原中,CO2还原到 CO 的总自由能变化决定了反应在一定电位下的可逆性,DFT 可预测目标产物的产率上限。
活化能(Activation Energy):催化反应的速率常由能垒高度决定。DFT 可结合过渡态搜索方法(如NEB)找到反应的过渡态,总能量比初态高出的部分即为活化能 Ea。活化能越低,反应动力学越快。
实验上,反应速率常服从阿伦尼乌斯关系 k=A⋅e–EaRT。DFT 还可用于微观动力学建模,通过计算各步的自由能和活化能来预测整体反应速率和选择性。
DFT 在催化剂设计中通常作为高通量筛选与描述符分析的工具,通过计算大量材料的关键能量指标,快速缩小候选范围。其常用策略包括:
描述符与火山图: 利用吸附能或相关电子结构参数(如过渡金属的d带中心)作为活性描述符。
按照萨巴蒂埃原理,可构建“火山曲线”:在横轴绘制某中间体的吸附自由能,纵轴为反应速率(或过电位等),大部分反应活性呈火山形趋势,即吸附能过强或过弱都降低活性。
通过DFT计算可确定描述符最佳区间。例如,在氢析氧或析氢反应中,*H 的吸附能零点处通常对应最高活性;在ORR中,*OH 吸附能接近峰顶时得到最低过电位。
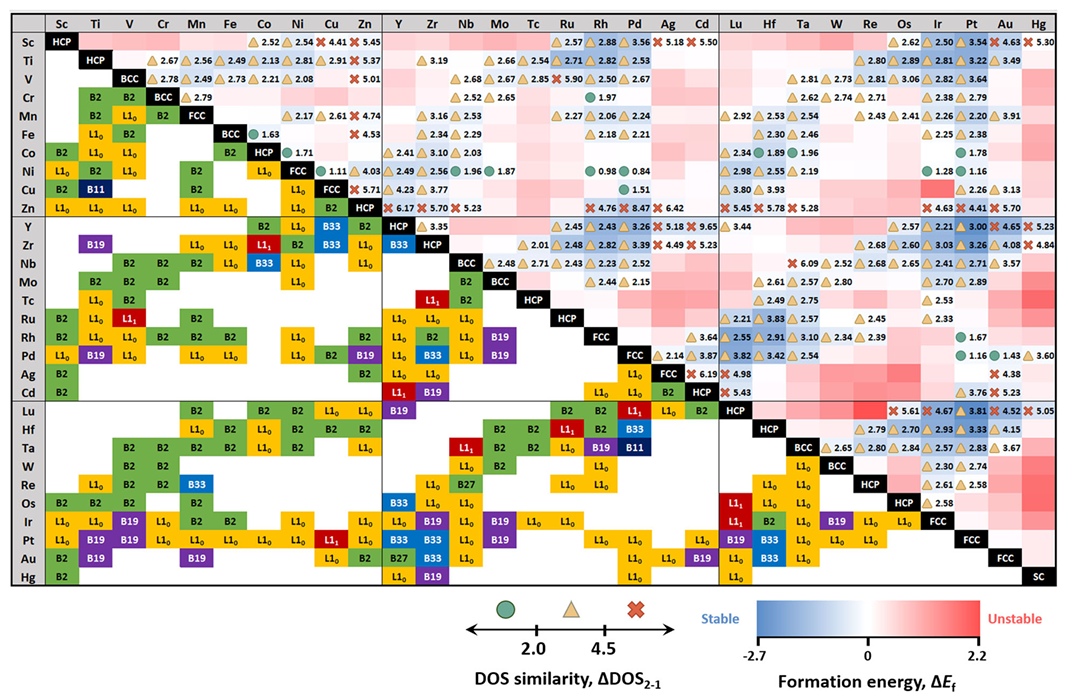
高通量计算: 结合自动化工作流程,利用DFT并行计算海量化合物(如多种合金、掺杂材料、二维材料等)的能量与吸附能。常见做法是在母体结构上随机替换或加掺新元素,几千、几万种构型通过超级计算机筛选。
如Yeo等人使用DFT筛选了4350种双金属合金结构,用电子态密度模式作为筛选描述符,最终提出了8个替代Pd催化剂的候选,并在实验中验证其中Ni61Pt39等表现优异,催化H2O2的效率比商业Pd高出9.5倍。
DOI:10.1038/s41524-021-00605-6
材料结构优化: DFT能够探究催化剂的结构和组成对活性的影响,指导掺杂、合金化、表面取向等改性策略。
例如,通过DFT计算不同掺杂元素对吸附能的调控效果,可预测何种掺杂最有利;利用DFT确定催化剂表面的活性位点、析氧析氢的有利吸附位点等。
对于新型催化剂体系,如碳负载单原子催化剂(SAC)或异质界面,DFT可用于评估不同金属原子或邻域的稳定性和活性,为实验合成提供靶标。
1. 确定反应和机理: 明确目标反应(如HER、OER、CO2RR等)及其可能的反应路径和关键中间体。
2. 构建候选材料库: 根据催化剂类型,选择不同金属、合金、氧化物、单原子位点等作为备选材料。例如,可从材料数据库筛选或自建化合物集。
3. 计算筛选描述符: 对每种候选催化剂利用DFT计算关键指标,如中间体吸附自由能、反应能、活化能等。也可计算电子结构参数(d带中心、功函数等)作为辅助描述符。
4. 分析与排序: 根据计算结果筛选出最优候选。常用方法包括绘制火山图、散点图等找到活性极值;利用机器学习或数据挖掘进一步加速排名。
5. 实验验证: 合成最优候选材料,进行电化学或催化反应测试。如电极测试当前密度-电势曲线、转化率、产物谱等,对比DFT预测的趋势。
6. 反馈优化: 根据实验结果调整计算模型(如考虑动力学、溶剂效应等),并可设计新的候选以进一步优化。
此流程强调DFT预测与实验验证的迭代协同:DFT提供指导候选材料方向,实验提供真实反馈,再由计算解释实验现象并指导下一轮设计。
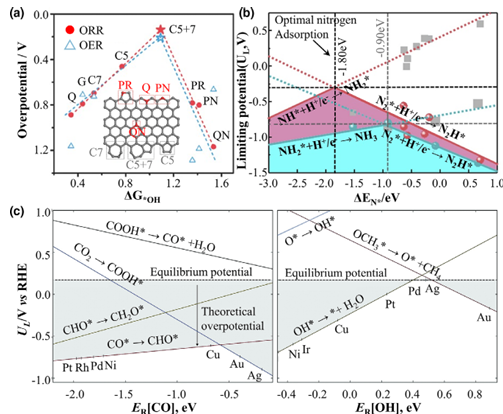
1. 电化学催化案例(如ORR、HER、CO2RR、NOX还原等): 以氧还原反应为例,Nørskov 等利用DFT指出OH吸附能是ORR活性的主要描述符,并提出了OER/ORR火山关系;此后大量研究在Fe–N–C、Pt–Co、Pt–Ni合金等体系中都证明了这一规律。
此外,对于二氧化碳还原反应,CO和COOH的吸附能被用来预测产物分布;对于氮气还原反应,由于N≡N键极难断裂,其NNH中间体的吸附能成为活性的关键描述符。
实验上,如某些研究通过DFT筛选预测出活性位点或合金组成,随后制备出的材料的选择性和活性与计算预言相符。
2. 热催化/光催化案例: 在热催化(如Fischer–Tropsch合成、NH3合成)和光催化(水分解、CO2光还原)领域,也有众多DFT应用。
例如,DFT可用于预测催化剂表面如何促进中间体键断裂与形成,判断氧化物上界面载流子能级对光催化活性的影响。
诸如通过DFT计算分析载流子分布和活化路径,指导设计合金或掺杂的热催化剂;或预测光催化半导体的带结构、表面吸附情况帮助选择合适材料。
这些案例多数证明,理论计算与实验协同极大提升了催化剂筛选的效率和材料设计的科学性。
对于不具备理论背景的实验催化研究者而言,DFT提供了强大的第一性原理指导能力:通过对原子级反应过程的能量计算,DFT能够在材料选择和结构设计上给出直观的指导意见,从而显著减少盲目实验量。
DFT计算所得的反应热力学能量、动力学能垒和吸附能等指标,与实际催化活性密切相关;利用这些指标构建的描述符(如吸附自由能、d带中心等)和火山图理论,已经在多种催化反应中得到了验证。
未来,随着计算能力和算法的发展,DFT与数据驱动方法(如机器学习)的结合将进一步加速催化材料设计的步伐,使实验与计算协同成为新常态,助力发现更高效、更廉价的催化剂。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!