

COHP(晶体轨道哈密顿布居)基于DFT量化原子间轨道相互作用的能量贡献,负值表成键,正值表反键,ICOHP积分值衡量键强度。
LOBSTER程序通过投影平面波数据实现COHP分析,应用于合金磁性、催化活性优化(如NiFe羟基氧化物中Mo掺杂弱化Fe-O键,激活晶格氧)。
未来结合机器学习加速筛选,开发动态COHP追踪键演变,多维分析(结合ELF、声子谱)揭示键合机制,推动材料设计从原子尺度到宏观性能的精准调控。


COHP的定义与基本原理
COHP(Crystal Orbital Hamilton Populations)是一种基于密度泛函理论(DFT)的化学键分析工具,通过量化相邻原子间轨道对相互作用的能量贡献,揭示材料的成键与反键特性。
其核心思想是将带结构能量分解为原子轨道对的相互作用,形成一种“键权重”态密度(DOS)。
晶体轨道重叠布居(COOP)与晶体轨道哈密顿布居(COHP)是分析化学键特性的两种重要方法,二者在理论基础和物理意义上存在显著区别:COOP 基于轨道重叠矩阵分析电子在原子间的分布情况,而COHP 则通过哈密顿矩阵元素考察能量分布,能够更直接地关联键的强度。
在符号约定上,COHP 的负值表示成键贡献(能量稳定化),正值表示反键贡献(能量不稳定化),为符合直观理解,通常绘制 “-COHP” 曲线,使正值对应成键作用,负值对应反键作用。
通过积分至费米能级的 COHP 值(ICOHP)可定量评估键的强度,ICOHP 绝对值越大,表明成键或反键作用越强,为键合特性提供量化指标。
COHP 的核心优势在于将电子结构的能量信息与化学键的成键特性相结合,弥补了传统态密度(DOS)仅反映电子能量分布位置而无法直接揭示键合本质的不足。
例如,在过渡金属氧化物中,COHP 分析可精准揭示金属 – 氧键的成键状态:低能区的负 COHP(对应成键)表明电子在金属与氧之间的有效共享,增强键的稳定性;而高能区的正 COHP(对应反键)则反映轨道相互作用的能量不稳定化,可能影响材料的催化活性。
这种能量与键合的关联分析,使 COHP 成为研究复杂体系中化学键强度、电子离域效应及反应活性的关键工具,尤其在催化材料设计、合金相稳定性分析等领域具有重要应用价值,能够从原子尺度阐明轨道相互作用对材料宏观性能的影响机制。
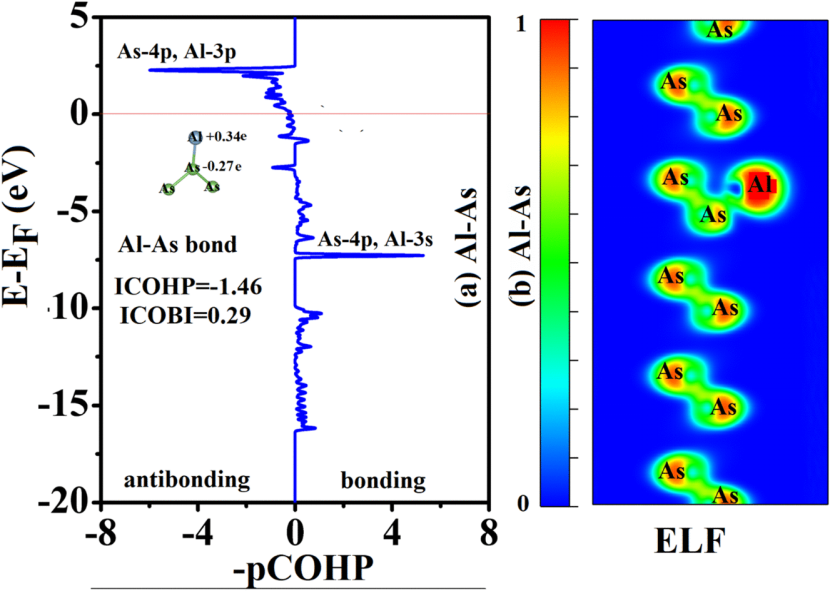
DOI:10.1039/d5ra00232j
DFT计算中的COHP
发展历程
COHP(晶体轨道哈密顿布居)理论的发展历程体现了计算化学方法从理论提出到技术适配再到功能扩展的持续演进。
1993 年,Dronskowski 与 Blöchl 首次提出 COHP 理论,最初基于紧束缚 LMTO-ASA 方法,针对局域轨道基组设计,为定量分析晶体中原子间成键与反键轨道的能量贡献奠定了基础。
然而,早期方法受限于基组类型,在平面波密度泛函理论(DFT)广泛应用的背景下,2011 年后随着 LOBSTER 程序的开发,这一局限被突破 —— 该程序通过投影平面波基组的波函数,实现了从 VASP、Quantum ESPRESSO 等主流 DFT 软件中提取 COHP 数据,解决了基组依赖性问题,推动 COHP 分析与平面波 DFT 框架的深度融合,极大拓展了其在实际材料体系中的适用性。
进入现代,LOBSTER 程序的功能进一步扩展,不仅支持自旋极化 COHP 分析(适用于磁性材料中自旋相关键合特性的研究),还实现了 k 空间依赖的 COHP 计算(可结合能带结构解析不同动量空间下的轨道相互作用),同时集成原子电荷计算功能,形成多维度的电子结构分析工具。
这些技术进步使 COHP 从最初的局域轨道基组理论发展成为兼容多种 DFT 计算框架、适用于复杂材料体系的普适性方法,在合金相稳定性、催化活性位点电子结构解析、磁性材料成键机制等研究中发挥关键作用,标志着从理论提出到工程化应用的重要跨越,为原子尺度化学键的定量分析提供了高效且可靠的技术路径。
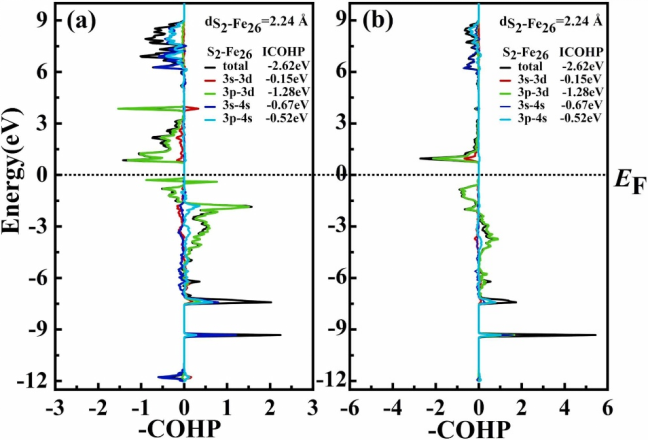
DOI:10.1016/j.colsurfa.2024.134256
计算流程
基于密度泛函理论(DFT)的 COHP(晶体轨道哈密顿布居)分析需遵循严谨的计算流程,主要包括结构优化、自洽计算、LOBSTER 输入设置及后处理分析四个关键步骤。
首先,使用 VASP 软件进行几何结构优化,通过迭代计算使能量收敛精度达到 EDIFF≤1e-6 eV,同时设置合适的能量截止参数(如 ENCUT≥500 eV)以确保平面波基组对电子波函数的准确描述,为后续电子结构计算奠定稳定的几何基础。
在自洽场(SCF)计算阶段,需启用 LORBIT=11 参数以记录投影波函数信息,生成包含轨道投影数据的 WAVECAR 和电荷密度文件 CHGCAR,这些数据是后续 LOBSTER 程序提取 COHP 信息的核心输入。
随后,通过编写 LOBSTER 输入文件(lobsterin)指定具体的分析对象,例如通过 “cohpBetween Atom1 3p 3d and Atom2 4s 4p” 语句定义特定原子对(Atom1 与 Atom2)及其参与成键的轨道(如 Atom1 的 3p、3d 轨道与 Atom2 的 4s、4p 轨道),实现对目标原子间轨道相互作用的定向分析。
最后进入后处理阶段,LOBSTER 程序基于输入参数计算并输出 pCOHP(投影晶体轨道哈密顿布居)曲线及积分值 ICOHP(积分至费米能级的 COHP 值),结合态密度(DOS)图,可直观解析不同能量区间内轨道的成键(负值对应成键贡献,通过绘制 “-COHP” 曲线使正值对应成键)或反键状态,以及键合强度(ICOHP 绝对值越大,键合作用越强)。
这一流程通过 VASP 与 LOBSTER 的协同,实现了从原子结构优化到化学键能量贡献的定量分析,为复杂体系中轨道相互作用的精准解析提供了标准化技术路径。
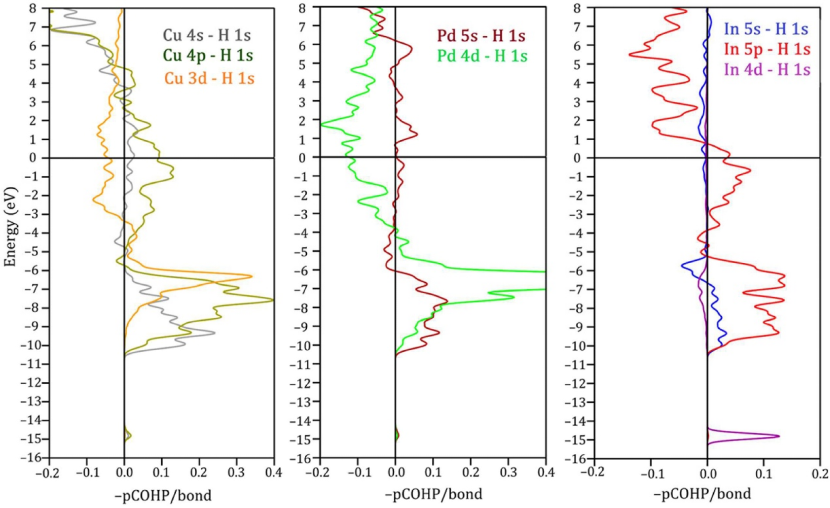
DOI:10.1016/j.ijhydene.2023.05.275
典型应用场景
COHP(晶体轨道哈密顿布居)分析在材料科学与催化领域的典型应用场景展现了其在原子尺度定量解析化学键特性的独特优势。
在过渡金属合金研究中,通过 COHP 可深入剖析 Fe-Fe 等金属键的磁性耦合机制:反键态电子的能量分布与占据情况直接影响原子磁矩的大小及相互作用,例如反键轨道的高电子占据可能削弱磁性耦合,而成键态的强轨道重叠则增强磁矩协同,为高性能磁性材料设计提供电子结构层面的理论依据。
在催化剂设计领域,COHP 的积分值 ICOHP 成为关键量化指标:以钙钛矿型催化剂为例,金属 – 氧键的 ICOHP 值与氧空位形成能密切相关 ——ICOHP 绝对值越大,键合越强,氧空位形成能越高,反之则更易形成氧空位以激活催化活性位点。
通过调控金属阳离子的 d 轨道与氧阴离子的 p 轨道相互作用,可精准预测并优化材料的氧还原或氧化反应活性,加速新型高效催化剂的开发进程。
在储氢材料开发中,COHP 分析用于评估氢原子与宿主材料(如金属氢化物、配位框架)的键合强度:通过计算 H 原子与金属原子间的成键 / 反键轨道能量贡献,结合 ICOHP 值可定量筛选具有适中键合强度的储氢材料 —— 键合过强导致释氢困难,过弱则影响储氢容量,而 COHP 提供的能量分布细节为平衡储氢稳定性与可逆性提供了关键参数。
这些应用场景共同体现了 COHP 方法从基础电子结构分析到实际材料性能调控的桥梁作用,为复杂体系的化学键工程提供了精准的理论工具。
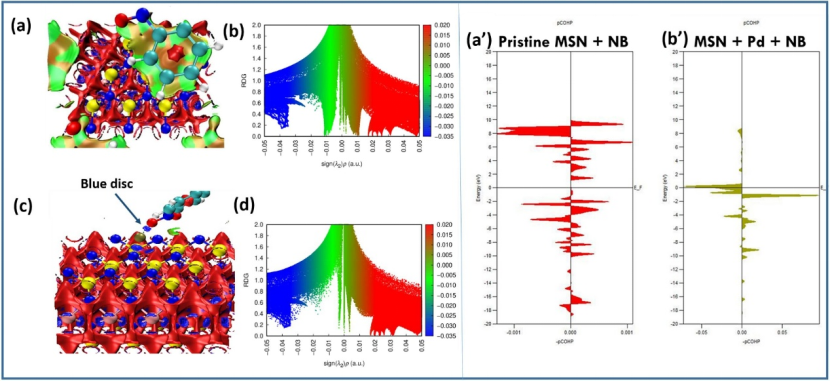
DOI:10.1016/j.molliq.2025.127310
实例分析:COHP应用
在《Activating lattice oxygen in NiFe-based (oxy) hydroxide for water electrolysis》(Nature Communications, 2022)的研究中,为揭示 NiFe 羟基氧化物中晶格氧的活化机制以优化电解水催化性能,研究者采用晶体轨道哈密顿布居(COHP)分析方法,结合密度泛函理论(DFT)展开深入研究。
首先通过 VASP 软件进行结构优化与电子结构计算,使用 PAW 赝势和 PBE 泛函,确保几何结构与电子分布的准确描述,随后借助 LOBSTER 程序提取 Ni-O 和 Fe-O 键的 – COHP 曲线及积分值 ICOHP,定量评估金属 – 氧键的强度与成键特性。
COHP 分析结果显示,Ni-O 键在 – 2 eV 至费米能级区间呈现显著的成键贡献(-COHP 曲线正值),表明该键具有较高稳定性;而 Fe-O 键在相同能量区间以反键贡献为主(-COHP 曲线负值),键合作用相对较弱。
当引入 Mo 掺杂后,Fe-O 键的反键区能量明显降低,-COHP 曲线中反键区域面积显著减少,同时 ICOHP 绝对值从 NiFe 体系的 – 1.8 eV 减小至 MoNiFe 体系的 – 1.2 eV,表明 Mo 掺杂有效弱化了 Fe-O 键。
这种键强度的调控使晶格氧的电子环境发生改变,原本受强键束缚的氧原子因 Fe-O 键弱化而更容易被激活,参与电解水反应中的关键中间体吸附与氧析出步骤。
该研究通过 – COHP 曲线的直观对比与 ICOHP 值的定量分析,从电子结构层面阐明了 “金属 – 氧键强度调控影响晶格氧活性” 的内在机制:反键区域的减少与键合强度的降低,本质上是 Mo 掺杂诱导的轨道杂化变化 ——Mo 的 d 轨道与 Fe 的 d 轨道及 O 的 p 轨道形成新的成键组合,改变了电子在金属 – 氧界面的分布,使氧原子的电荷密度增加,活性位点的氧化还原能力提升。
这一发现打破了传统催化剂设计中依赖活性金属位点的局限,提出通过调控晶格氧配位环境(即金属 – 氧键的成键 / 反键状态)激活氧自身反应性的新策略,为高性能电解水催化剂的设计提供了全新视角。
该实例充分展现了 COHP 分析在催化材料研究中的独特优势:通过将轨道相互作用的能量分布转化为可量化的键强度参数(ICOHP),结合可视化的 – COHP 曲线,精准定位活性位点的电子结构特征,从而建立“掺杂 – 轨道耦合 – 键强度 – 催化活性” 的内在关联。
这种从原子尺度到宏观性能的跨尺度分析,不仅深化了对晶格氧活化机制的认识,更验证了 COHP 方法在指导复杂金属氧化物催化剂设计中的实用性,为推动电解水技术的工业化应用奠定了理论基础。

DOI:10.1038/s41467-022-29875-4
总结
COHP 作为密度泛函理论(DFT)计算中定量解析材料化学键的核心后处理工具,已在电子结构分析、催化机制阐释及材料性能优化等领域展现出重要价值。
未来发展方向聚焦于三方面突破:一是通过与机器学习算法结合,构建高通量计算框架,快速挖掘 ICOHP(积分晶体轨道哈密顿布居)与材料力学、催化、磁性等性能的关联规律,加速新型功能材料的筛选与设计;
二是开发时间分辨 COHP 技术,捕捉催化反应、相变过程中化学键的动态演化(如键的断裂与形成路径),为揭示复杂动态体系的反应机理提供实时能量分布视角;
三是推动多维分析扩展,将 COHP 与电荷密度分析(如 ELF)、声子谱等多尺度表征手段结合,从电子结构、电荷分布及晶格振动等维度协同揭示键合机制,构建更完整的材料结构 – 性能关系模型。
这些方法创新将进一步夯实 COHP 在理论化学与材料科学中的桥梁作用,推动从原子尺度电子结构分析到宏观性能调控的理论设计闭环,为精准化、智能化材料研发提供更高效的工具支撑。
写在最后
热门半导体计算方法在MS半导体课程中均有讲解。
本次课程将由华算科技技术部资深专家、经验丰富的杨站长带大家学习杂化钙钛矿 + 二维材料 + 异质结构 + 气敏半导体的电子结构与晶格动力学性质计算。
