


同步辐射XAFS在单原子领域中的应用
什么是同步辐射R空间?
如何对XAFS数据预处理与拟合分析?
精选干货|同步辐射PDF基础知识及经典应用分析!
如图1所示实验布局示意图,主要实验挑战在于:1)获得可精确调谐的单色X射线源;2)高质量的X射线强度探测器。
多数实验使用同步辐射源提供全波段X射线,通过基于布拉格衍射的硅双晶单色器选择特定能量。该单色器由两个晶面间距相同的晶体组成:第一晶体旋转选择衍射能量,第二晶体对衍射束进行二次衍射使其与原光束平行,从而保证不同能量单色光最终聚焦于样品同一位置。
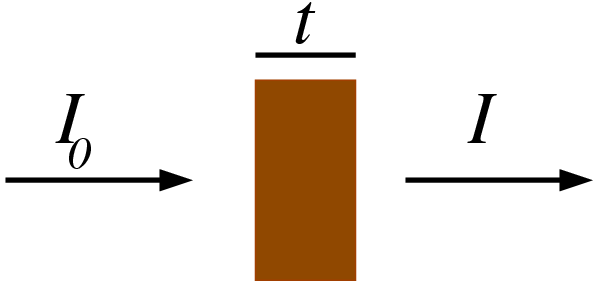
图1:X射线吸收测量原理示意图。一束强度为I₀的单色X射线入射穿过厚度为t的样品后,透射光束强度衰减为I。
对XAFS至关重要的单色器特性包括能量分辨率、稳定性和重复性。硅单色器在10keV能量下可达~1eV分辨率,完全满足XAFS需求。由于布拉格角调节精度需达10-4°(微小角度变化即引起显著能量偏移),加之温度漂移会影响能量稳定性,这些因素虽在现代XAFS专用光束线上已基本解决,但仍需注意。
基于布拉格衍射的单色器不仅会选出基频能量,还会产生谐波(能量整数倍)。这些高能谐波在吸收边以上区域不会被样品有效吸收,可能导致难以诊断的数据异常(如显著“毛刺“和噪声)。
消除谐波主要有两种策略:一是使单色器双晶略微失谐(通过压电晶体微调第二晶体位置),这会大幅抑制高次谐波强度;二是在光路中加入X射线反射镜(对基频反射效率远高于谐波)。理想情况下可组合使用这两种方法。
透射测量中检测I0和I的线性探测器(如充入惰性气体的电离室)技术成熟。但需注意:产生的pA级电流需经放大传输,放大器在高低增益区可能存在非线性,且存在暗电流干扰。荧光测量还可选用其他探测器,但线性度问题更为突出。
对于均匀样品,只要保证光束准直和样品无针孔,透射测量通常能轻松获得精确数据。荧光测量虽也能达到10-3噪声水平,但对低浓度样品更具挑战性。
高浓度样品(待测元素>10%)应采用透射法。样品厚度需满足μt≈2.5(吸收边以上)或Δμ(E)t≈1(边阶跃)。例如铁金属需7μm厚度,金属氧化物通常数十微米,稀溶液则需毫米级厚度。
此外要求样品均匀无孔洞,粉末颗粒尺寸不得超过吸收长度。满足这些条件(有时具有挑战性)的透射测量能获得优质数据,适合标准样品测试。
对于厚样品或低浓度样品(最低至ppm级),荧光检测是优选方案。荧光XAFS测量通过监测样品发出的特征X射线荧光来实现。
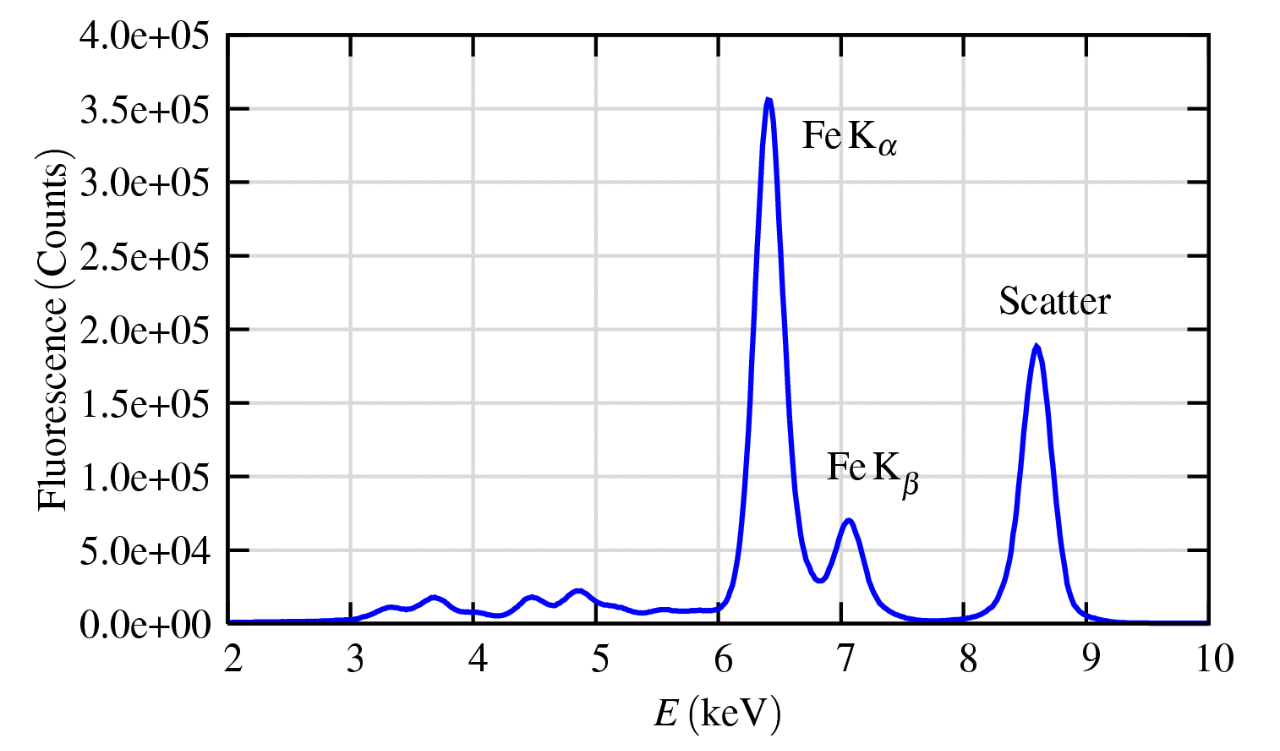
图2:富铁矿物(橄榄石)的X射线荧光光谱图,其中显示了Fe的Kα和Kβ发射谱线,以及弹性(和近弹性)散射峰。在较低能量区域还可以观察到Ca、Ti和V的特征峰。
样品中的目标荧光谱线、其他元素的荧光谱线,以及弹性和非弹性(康普顿)散射的X射线会同时出现。
图2展示了一个典型的荧光光谱,其中包含Fe的Kα和Kβ荧光谱线、弹性散射峰(与康普顿散射无法分辨),以及来自Ca、Ti和V的荧光谱线。在许多情况下,散射或其他元素的荧光谱线会主导整个荧光光谱。
进行高质量的荧光XAFS测量时,有两个主要考量因素:探测器收集的立体角和荧光谱线的能量分辨率。
立体角的需求容易理解——荧光是各向同性发射的,我们希望尽可能多地收集可用信号。而弹性和非弹性(如康普顿)散射的X射线并非各向同性,因为同步辐射产生的X射线在加速器平面内具有偏振性。
这种偏振特性意味着,在水平面上与入射光束呈90°的方向,弹性散射会被大幅抑制。因此,荧光探测器通常被放置在垂直于入射光束的位置。

图3:“Z-1”过滤器对实测荧光光谱的影响。在样品与探测器之间放置Mn过滤器时,该过滤器会吸收大部分散射峰,同时允许Fe的Kα发射峰通过。对于散射峰占主导的样品,此类过滤器可显著提升信噪比。
荧光探测器的能量分辨率至关重要,因为它能基于能量差异区分信号,从而抑制散射X射线和其他元素的荧光谱线,相对增强目标荧光谱线的强度。
这可以降低背景噪声,提高信噪比。能量甄别可通过物理方式(在X射线到达探测器前过滤)或电子方式(探测后处理)实现。
一种常用的物理过滤方法示例是:在含Fe样品与荧光探测器之间放置富Mn材料。由于Mn的K吸收边,Mn会优先吸收散射峰,同时允许Fe的Kα谱线通过(见图3)。这种简单的过滤器可用于无固有能量分辨率的探测器(如电离室)。
为避免过滤器自身再辐射,通常会使用一组狭缝(索勒狭缝)优先收集样品发射的信号并阻挡来自过滤器的辐射(如图3所示)。这种配置在信号以散射为主且样品浓度在数百ppm及以上时尤为有效。
能量甄别也可通过电子方式对探测到的X射线荧光光谱进行后处理实现。常见的方法是使用固态Si或Ge探测器,其能量分辨率可达约200 eV甚至更高,通常运行时的分辨率优于1 keV。图3所示的光谱即由此类Ge固态探测器采集。
这类探测器的显著优势是能测量完整的X射线荧光光谱,这本身对识别和量化样品中其他元素的浓度非常有用。由于可以完全剔除荧光光谱中的干扰部分,这类探测器可用于低至ppm级别的XAFS测量。
尽管固态探测器有许多优点,但也存在一些缺点:
✅死时间问题:电子能量甄别需要一定时间,这限制了可处理的总信号量。这类探测器的总计数率通常饱和在约105Hz左右。
当超过此速率时,探测器实际上无法记录所有荧光信号,部分时间处于“死”状态。因此,通常会并联使用十个(或更多)此类探测器,即便如此,这些探测器上的总入射强度限制仍可能影响XAFS测量的质量。
✅复杂性高:维护、设置和使用此类探测器的工作量远大于电离室。探测器通常需保持在液氮温度下,且能量甄别电子设备的“易用性”差异较大。此外,探测器和电子设备的价格也相当昂贵。
尽管存在这些缺点,固态探测器目前仍是XAFS测量的常用手段,尤其适用于稀薄或非均质样品。同时,探测器和电子设备本身也在持续改进中。
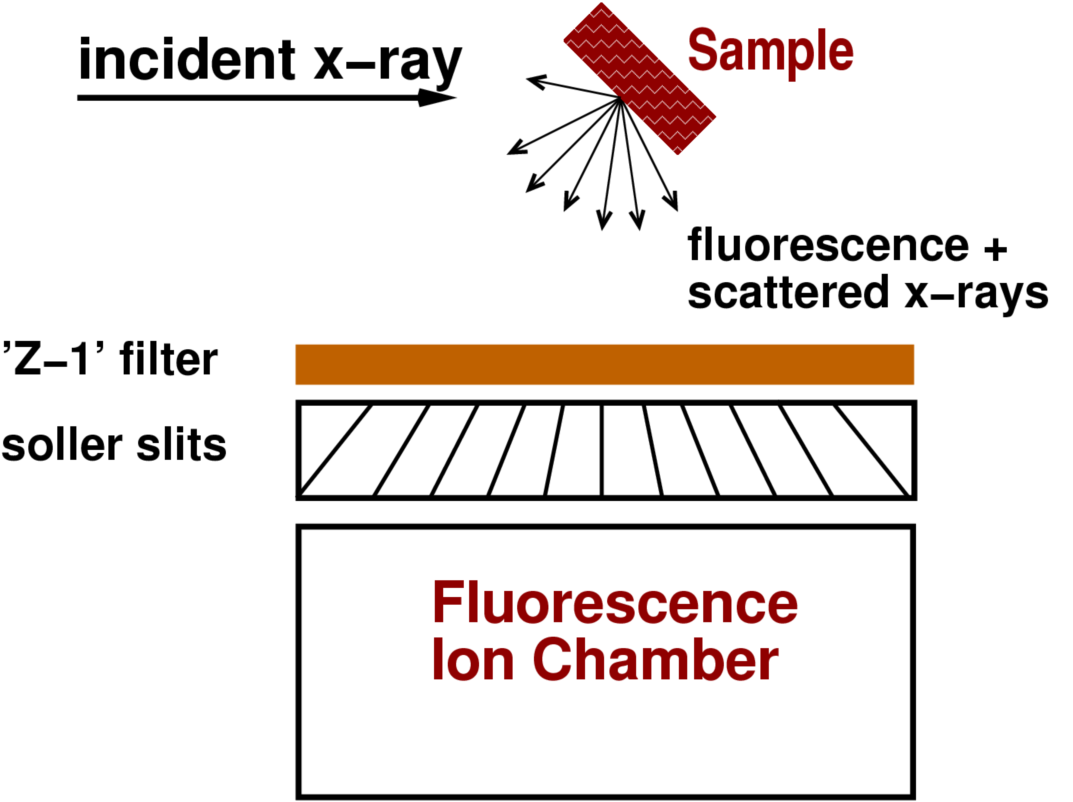
图4:”Z-1″过滤器在荧光光谱能量甄别中的实际应用示意图。置于样品与探测器之间的过滤器可吸收大部分散射峰,但其自身可能产生二次辐射。由于过滤器的辐射同样具有各向同性特性,一组指向样品的金属索勒狭缝可优先吸收来自过滤器的辐射。
关于X射线荧光测量的讨论,还需提及一个重要效应:自吸收。这一术语可能容易引起混淆。样品本身会吸收大量荧光X射线。例如,对于高Z基质(如氧化铅)中的低Z稀释元素(如硫),硫的荧光信号将严重衰减,实测信号由荧光X射线的逃逸深度决定。
但“自吸收”通常并非指上述情况,而是指样品的穿透深度主要由目标元素主导的情形。在最严重的自吸收情况下(如纯元素的厚样品),XAFS仅会改变X射线在样品中的穿透深度,但几乎所有X射线都会被目标元素吸收。
荧光X射线的逃逸深度通常远大于穿透深度,因此几乎所有被吸收的X射线都会产生荧光X射线。这会严重削弱XAFS振荡信号。对于极高浓度的样品,可能完全检测不到XAFS振荡!
前文我们提到,对于荧光模式测量的XAFS,其表达式为:
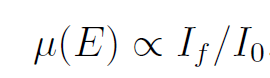
这一表述存在一定简化。荧光产生概率虽与吸收概率成正比,但实际测得的荧光强度需要穿过样品才能到达探测器。由于所有物质都会衰减X射线,荧光强度(进而XAFS振荡信号)会因这种自吸收效应而衰减。更准确的表述应考虑样品几何(参见图5):
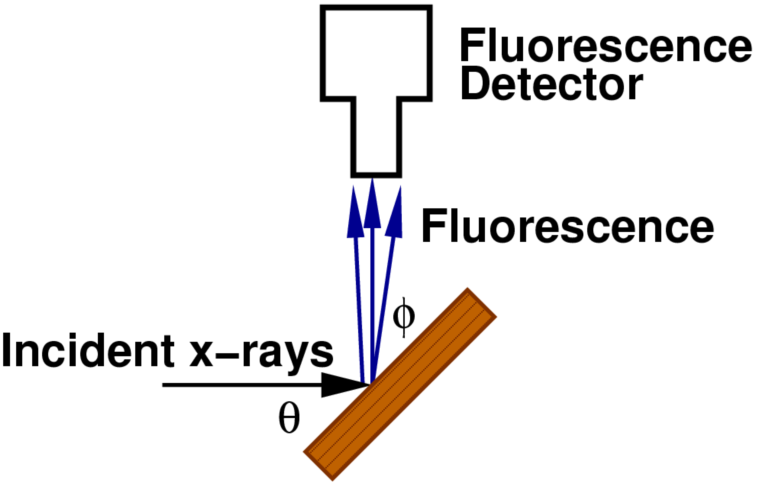
图5:荧光X射线吸收测量示意图,标注入射角θ和出射角φ。
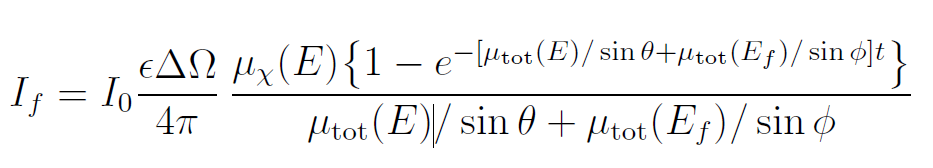
式中:ϵ为荧光效率,ΔΩ为探测器立体角,Ef为荧光X射线能量,θ 为入射角(入射X射线与样品表面夹角),ϕ为出射角(荧光X射线与样品表面夹角),μx(E)为目标元素吸收系数,μtot(E)为样品总吸收系数
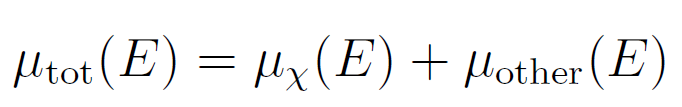
在实际XAFS测量中具有若干典型极限情况。首先是薄样品极限(μt≪1),此时 1−e−μt项可通过泰勒级数展开近似为:≈[μtot(E)/sinθ+μtot(Ef)/sinϕ]t
该近似式将与分母相消,从而得到:
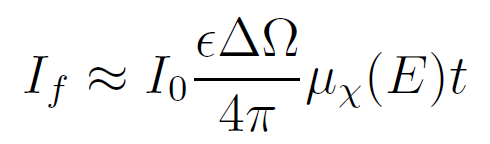
另一种情况是厚稀释样品极限,此时满足 μt≫1且 μx≪μ其他。在此条件下,指数项趋近于0,因此
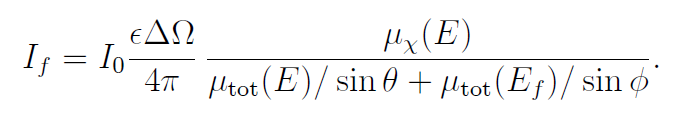
此时我们可以忽略μ总μ总的能量依赖性,从而:
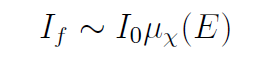
这两种极限情况(极薄或厚稀释样品)是荧光测量的最佳条件。
对于浓度较高的厚样品(μX∼μ其他,此时μX∼μtot,我们无法忽略μtot 的能量依赖性,必须对式中μtot (E)的振荡进行校正。如前所述,对于极高浓度样品,μtot (E)≈μX(E),可能导致XAFS信号完全消失。不过若自吸收效应不太严重,仍可通过上述方程进行校正。
对于高浓度厚样品,通过旋转样品使其与入射光束近乎垂直(即ϕ→0的掠射出极限),可有效降低自吸收效应。此时满足μtot (Ef)/sinϕ≫μ总(E)/sinθ,从而:
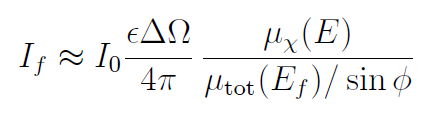
该操作可消除分母的能量依赖性。
在某些情况下,通过监测发射电子(包括俄歇电子和低能二次电子)的强度可进行XAFS测量。
由于材料中电子的逃逸深度通常小于1μm,这种测量方式比X射线荧光测量具有更高的表面灵敏度。电子产额测量最适用于金属或半导体样品(即样品需具备足够导电性,以补充发射电子并避免电荷积累)。基于这些原因,电子产额法在XAFS测量中并不常用。