

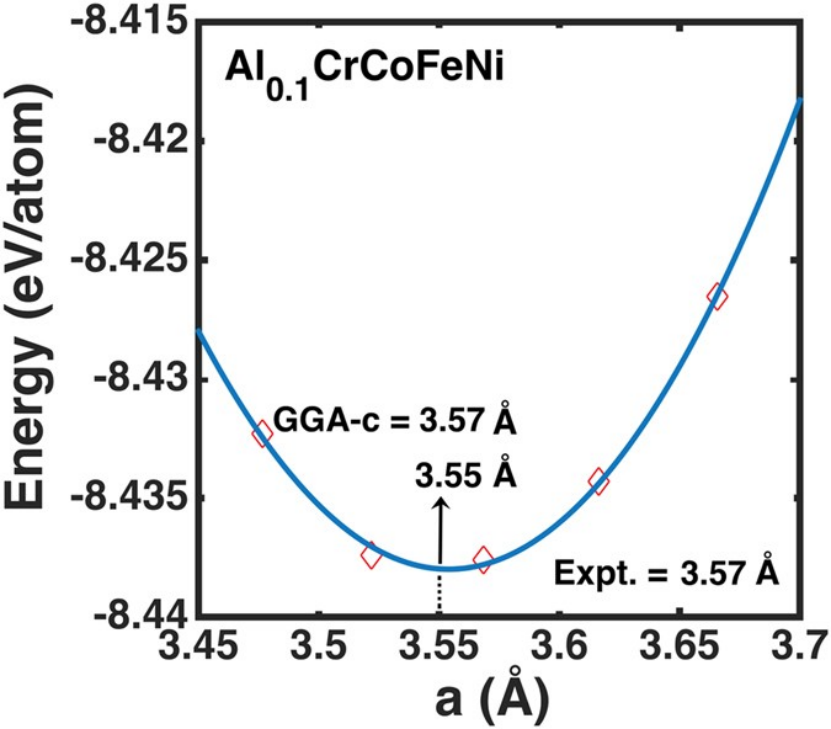

(Sci Rep 6, 31028 (2016). https://doi.org/10.1038/srep31028)
缺陷与掺杂结构:引入空位、间隙原子或异质原子后,优化局部原子环境。
例如,在二维Fe₂C₁₂材料中,通过缺陷工程优化碳原子排列,发现特定空位构型可显著提升材料的催化活性。
此类优化需结合声子谱分析,确保缺陷结构的动力学稳定性。
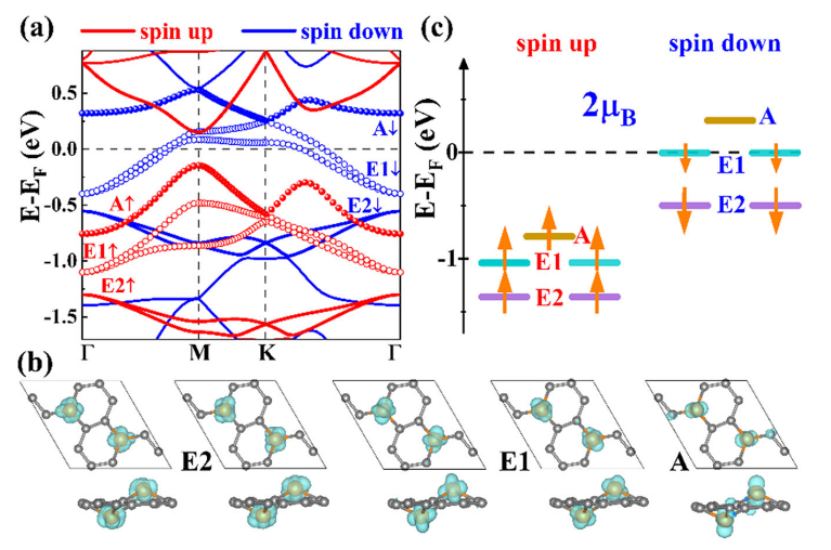
(https://doi.org/10.1038/s41699-021-00235-y)
界面与复合结构:在多相材料(如SiC/Al复合材料)中,优化界面原子的键合模式。
机器学习加速的结构优化方法通过预测界面分离能,快速筛选出具有高结合能的合金元素,从而降低计算成本。
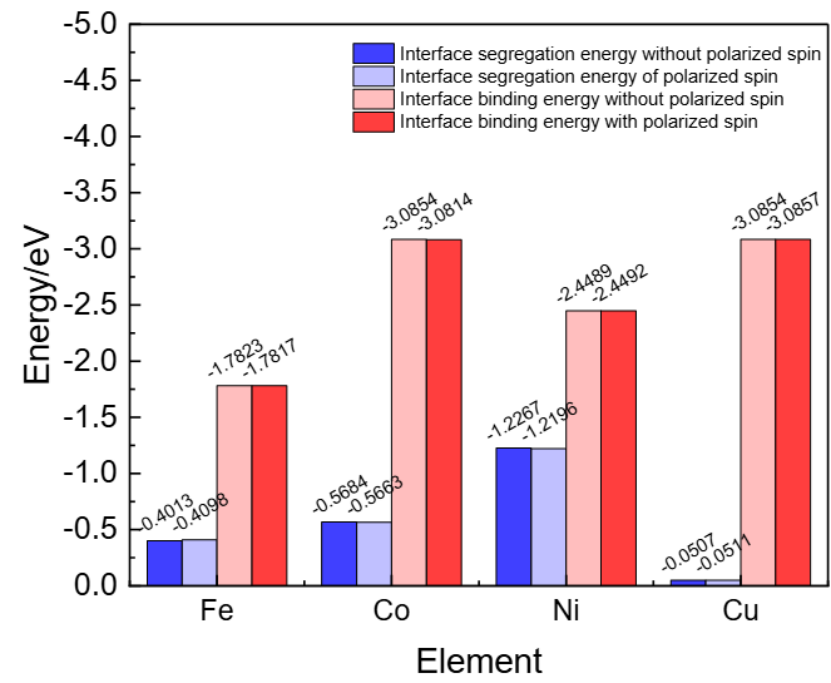
(https://doi.org/10.3390/ma17061322)
从势能面到优化算法
结构优化的数学本质是在高维势能面上寻找全局极小值。这一过程涉及以下关键步骤:
势能函数与原子间作用力
第一性原理计算中的势能面由量子力学方程(如Kohn-Sham方程)定义。以密度泛函理论(DFT)为例,总能量可表示为:
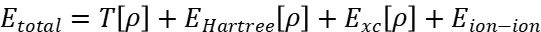
其中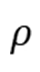 为电子密度,为交换关联能。原子间作用力通过Hellmann-Feynman定理计算:
为电子密度,为交换关联能。原子间作用力通过Hellmann-Feynman定理计算:
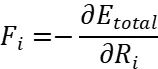
这些力驱动原子向势能面梯度下降方向移动。
优化算法的数学实现
最速下降法(Steepest Descent) :是一种经典的梯度下降优化方法,其基本思想是沿势能梯度的负方向,即原子所受合力的反方向,逐步调整原子位置,从而降低系统总能量。
该方法每一步的步长通常通过线搜索(line search)确定,以确保每一步都能有效降低能量。
尽管最速下降法实现简单、计算代价较小,适合初始构型离局部最小值较远的系统,但它的收敛速度较慢,特别是在高维势能面中,当接近极小点时常表现出“锯齿状”振荡行为,易陷入局部极小而难以进一步优化,因此通常用于早期粗优化,而非精细结构收敛阶段。
共轭梯度法(Conjugate Gradient, CG) :是一种改进的优化方法,适用于具有连续导数的多维函数最小化问题,特别适合处理二次型或近似为二次型的势能面。
与最速下降法不同,CG方法在每一步不再简单沿梯度方向移动,而是构造彼此“共轭”的搜索方向,避免在不同方向上反复来回,从而加快收敛速度。
在固体物理中,如硅晶体结构优化中,CG方法表现出良好的收敛性,一般只需几十步(如50次以内)即可将总能量收敛至极低误差范围(如1 meV/atom)。
它的高效性使其成为多种分子模拟和密度泛函计算程序中的默认选择之一。
BFGS算法:一种准牛顿方法,是一种在不显式计算Hessian矩阵(即二阶导数)的情况下高效逼近其逆的方法。
BFGS通过历史梯度与位移信息递推更新一个近似的Hessian逆矩阵,从而预测更优的搜索方向。
这种方法在处理非线性、多变量优化问题时具有出色的稳定性和快速收敛性能。其核心更新公式为:
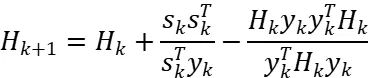
其中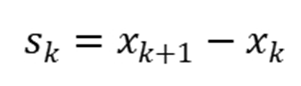 ,。BFGS在金属体系(如Fe、Co、Ni)中表现优异,因其能有效处理非光滑势能面。
,。BFGS在金属体系(如Fe、Co、Ni)中表现优异,因其能有效处理非光滑势能面。
收敛标准与精度控制
通常设定能量变化阈值(如1e-5 eV/atom)和最大力阈值(如0.01 eV/Å)作为收敛标准。
例如,在石墨烯缺陷优化中,当原子受力小于0.02 eV/Å时,认为结构达到稳态。
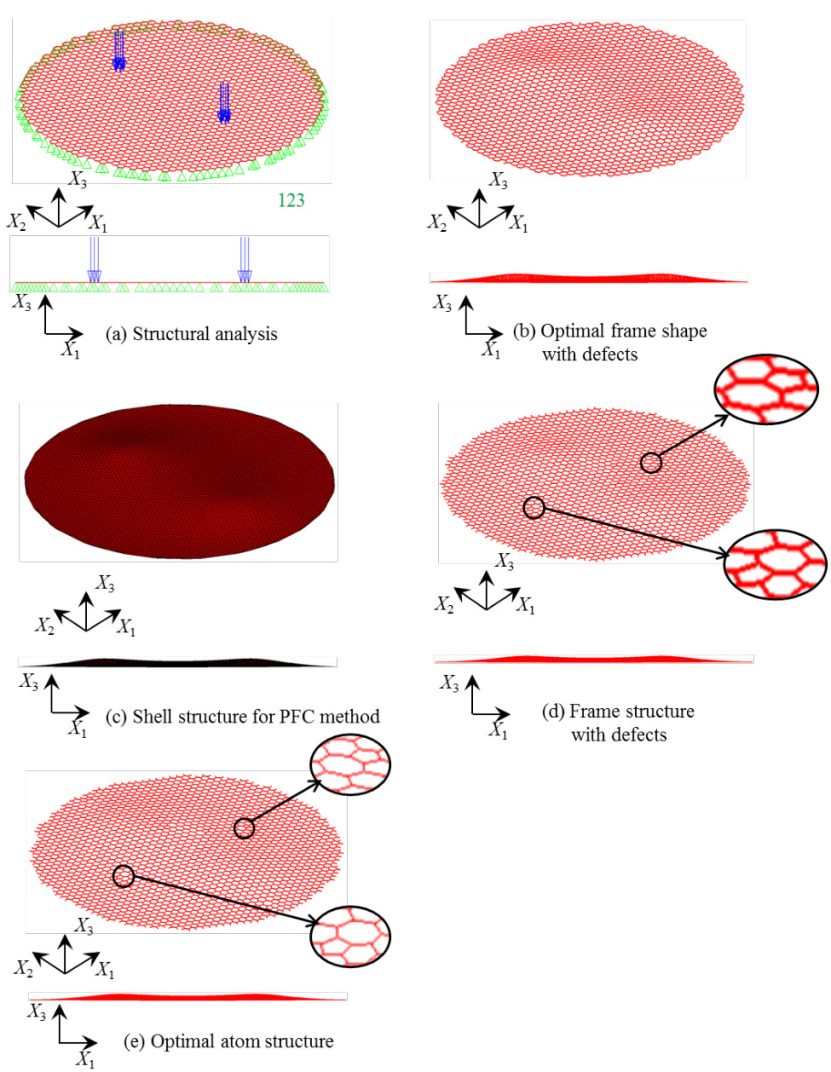
(DOI: 10.1299/transjsme.17-00077)
从硅晶体到石墨烯缺陷
硅晶体的晶格优化
在硅基光电子材料设计中,通过DFT计算优化晶格常数和带隙。
例如,某研究通过调整硅晶格参数,发现当晶格常数从5.43 Å压缩至5.38 Å时,带隙从1.1 eV增至1.3 eV,从而提升发光效率。
优化过程中,能量随晶格参数的变化曲线呈现抛物线型,极小值点对应平衡晶格常数。
石墨烯缺陷结构的原子级优化
单原子空位 :通过移除一个碳原子,局部形成三个悬键。
优化后,邻近原子发生弛豫,悬键重构为五元环和七元环(Stone-Wales缺陷),体系能量降低约4 eV。
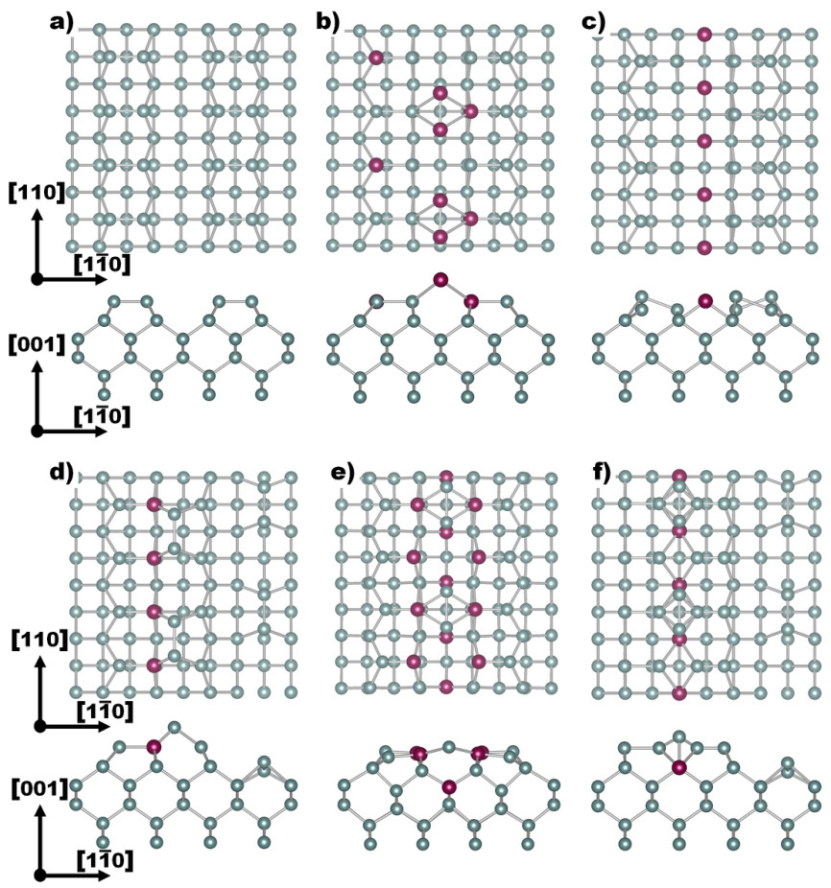
(DOI 10.1088/0953-8984/26/13/133001)
双原子空位 :两个相邻碳原子缺失导致更大的结构畸变。优化后形成两个五元环和一个八元环,电子局域化程度增强,显著影响导电性。
掺杂缺陷 :在石墨烯中引入氮原子后,优化其取代位置。计算表明,氮原子倾向于占据sp²杂化位点,导致费米能级上移0.2 eV,增强电化学活性。
界面结构的机器学习加速优化
在SiC/Al复合材料中,传统DFT计算需数周时间优化界面模型。
通过机器学习方法(如支持向量机)预测界面分离能,仅需1%的计算量即可筛选出最优掺杂元素(如Mg、Ti),使界面结合能提升15%。
挑战与未来方向
尽管结构优化方法已取得显著进展,仍面临以下挑战:
高维空间的计算复杂度
对于含有上千个原子的体系,例如金属合金、大分子或缺陷复杂的晶体结构,其势能面的维度非常高,传统优化算法(如最速下降法、共轭梯度法)在高维空间中容易陷入局部极小值,且每一步迭代所需的能量与力的计算也异常昂贵。
因此,逐步优化原子结构不仅效率低,而且在精度与可扩展性之间难以权衡。
为了解决这一难题,研究者提出了基于机器学习的势能函数(MLFFs),如高斯近似势(GAP)或神经网络势能(NNP),它们通过训练大量第一性原理数据,构建出可同时兼顾效率与精度的近似势能面,使得在高维体系中实现快速结构优化成为可能。
动态与温度效应
结构优化通常默认材料处于绝对零度(0 K)下的基态构型,然而实际中大多数材料工作于有限温度甚至非平衡环境中,如催化反应界面、电池充放电过程或应力加载条件下。
这种热扰动和动力学演化会显著改变体系的稳定结构和能量分布。因此,仅依赖静态优化方法并不能完全反映真实物理过程。
为了更全面评估材料行为,研究人员引入分子动力学(MD)模拟或蒙特卡洛(MC)方法,结合温度采样(如Berendsen或Langevin温控),可以获得热平衡态下的结构平均信息,并观察关键结构转变和弛豫路径,从而提升优化结果的实际可靠性。
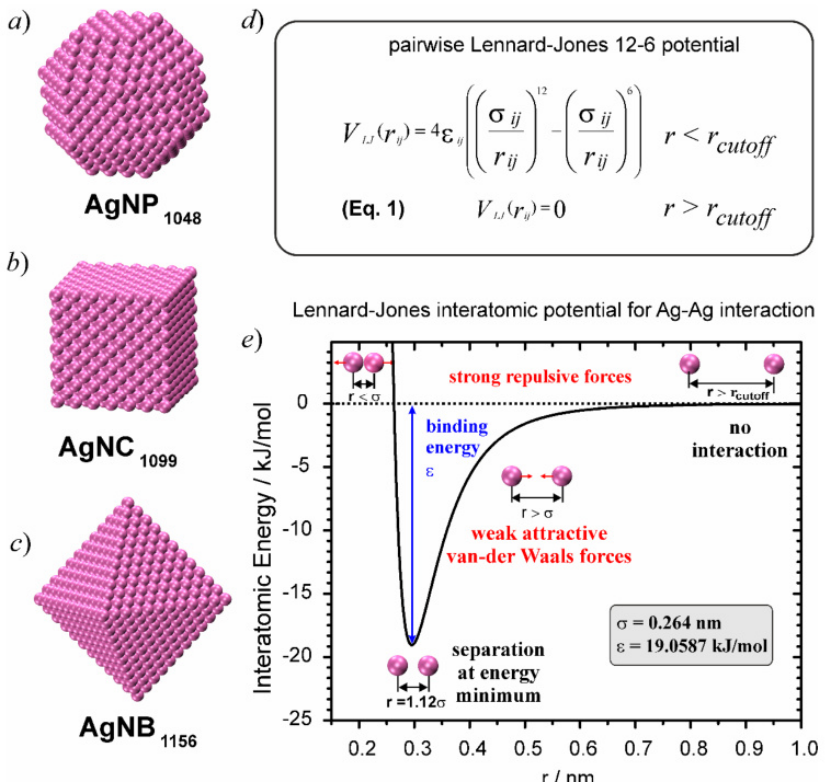
(DOI: 1026565/2220-637X-2017-29-02)
多尺度耦合
在涉及复杂材料或功能结构(如碳纳米管–石墨烯复合材料、电极多孔网络)中,优化不仅仅局限于原子尺度的构型调整,还需考虑更高尺度的介观结构,如孔隙分布、界面结构和宏观取向。
当前的单一尺度优化方法难以同时兼顾局部化学键合(如C–C键强度)与整体几何拓扑(如多孔网络的连通性),这限制了性能预测的准确性。
为此,多尺度建模成为研究热点。它通过将第一性原理计算与粗粒度模型(如有限元、晶粒演化模型)结合,实现从原子级别的构型到宏观功能表现的统一优化。
在实践中,这类方法可用于设计多孔电极、热界面材料等跨尺度性能主导系统,为先进材料开发提供新的策略。
通过上述分析可见,第一性原理中的结构优化不仅是调整原子位置的数值过程,更是理解材料本征性质、设计新型功能材料的核心工具。
从量子力学方程出发,结合高效算法与先进计算技术,科学家得以在原子尺度上“雕刻”材料,为能源、电子、催化等领域的突破提供理论基石。