

X 射线衍射(X-ray Diffraction,简称 XRD)技术作为一种强大的分析工具,在材料科学、地质学、化学、物理学以及生物医学等多个领域发挥着不可替代的作用。
不同测试技术与方法
广角 XRD
适用于大多数材料的物相分析和晶体结构分析。通过旋转样品和探测器,收集不同角度下的衍射数据,得到完整的 XRD 图谱。 小角 XRD
主要用于研究材料的微区结构和小尺度范围内的晶体结构变化。其测试角度范围较小,但可以提供更高的分辨率和灵敏度。
掠入射 XRD
掠入射 XRD 技术在材料科学中有广泛的应用,包括晶体生长、薄膜制备、界面化学、表面催化和生物材料等领域。通过调整 X 射线的入射角度和探测器位置,可以实现对样品表面层的精确分析。
原位 XRD
能够在样品制备和测试过程保持不变的情况下,实时监测材料的结构和性能变化。这对于研究材料的相变、应力松弛、热膨胀等现象具有重要意义。
X 射线衍射(XRD)应用实例 物相分析 不同气候带土壤粘粒矿物 XRD 物相分析
吉林农业大学姚乃慈等人以我国 7 个不同气候带下的森林土壤为研究对象,研究不同的影响因素,并使用 X 射线衍射分析方法,分析不同森林土壤粘粒矿物 XRD 物相特征,深入探讨在不同气候带下、受不同温度及降水影响的森林土壤的粘粒矿物的组成特点及含量变化。
寒温带(漠河)森林土壤粘粒矿物 XRD 图谱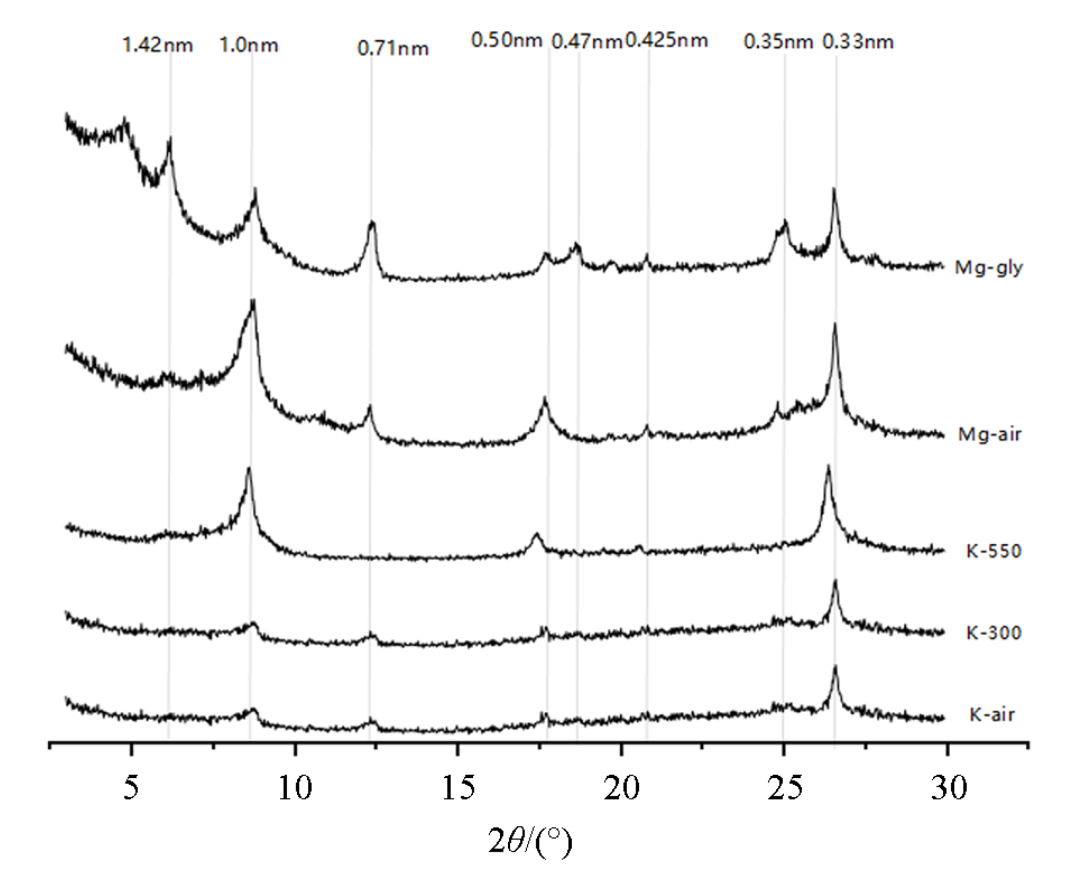

中温带、暖温带、北亚热带森林土壤粘粒矿物 XRD 图谱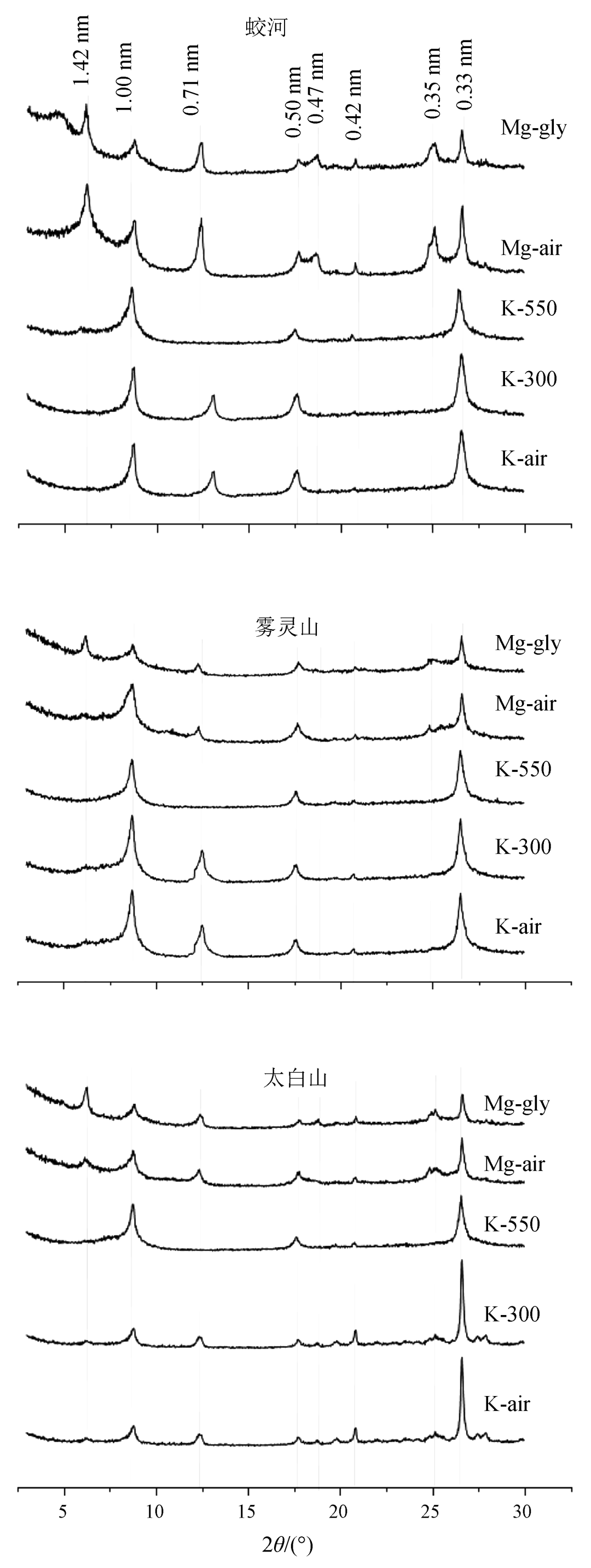
中亚热带、南亚热带、热带森林土壤粘粒矿物 XRD 图谱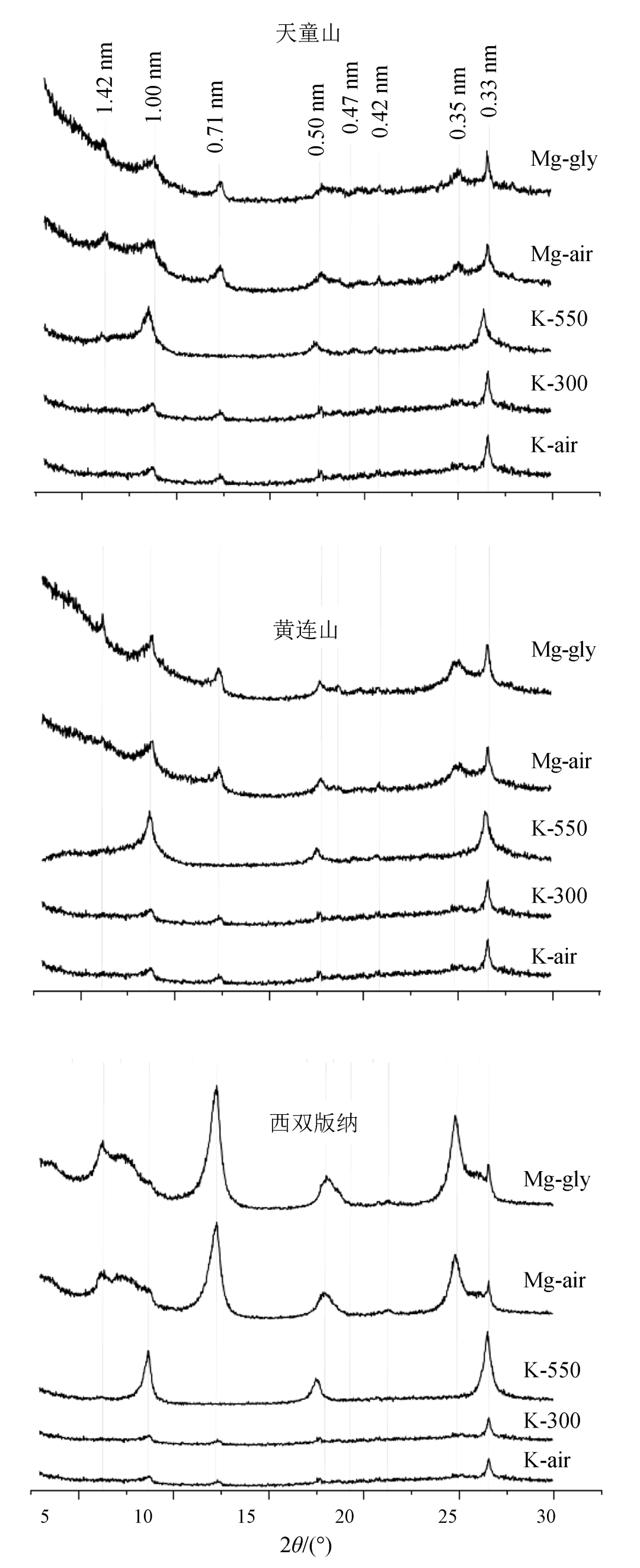
定量分析 XRD 定量分析蒙脱石含量及影响因素研究
根据 XRD 的原理可知,其实质是晶体衍射,衍射峰的强度与晶体的结构和晶体物质的含量有关:晶体结构完整、晶形较好,衍射强度越高;晶体结构残缺、晶形较差,衍射强度越低;晶体物质的含量越高衍射强度也就越强。 XRD 法定量分析中,常用的方法为基体清洗法(K 值法)和自清洗法(绝热法),这两种方法中各种矿物的 K 值可以在 PDF 卡片中查得。 董文辉等人以大量试验数据为基础,总结了生产实践中用 XRD 法精确定量蒙脱石含量的方法及影响因素。 在定量分析中,影响 X-射线衍射定量分析的因素很多,概括起来主要有三个方面: (1)样品状态造成的误差,如择优取向、颗粒效应、显微吸收、消光效应和结晶度等常常给定量分析结果带来较大误差; (2)强度测量软件的统计误差; (3)仪器带来的误差。
人工钠化蒙脱石谱图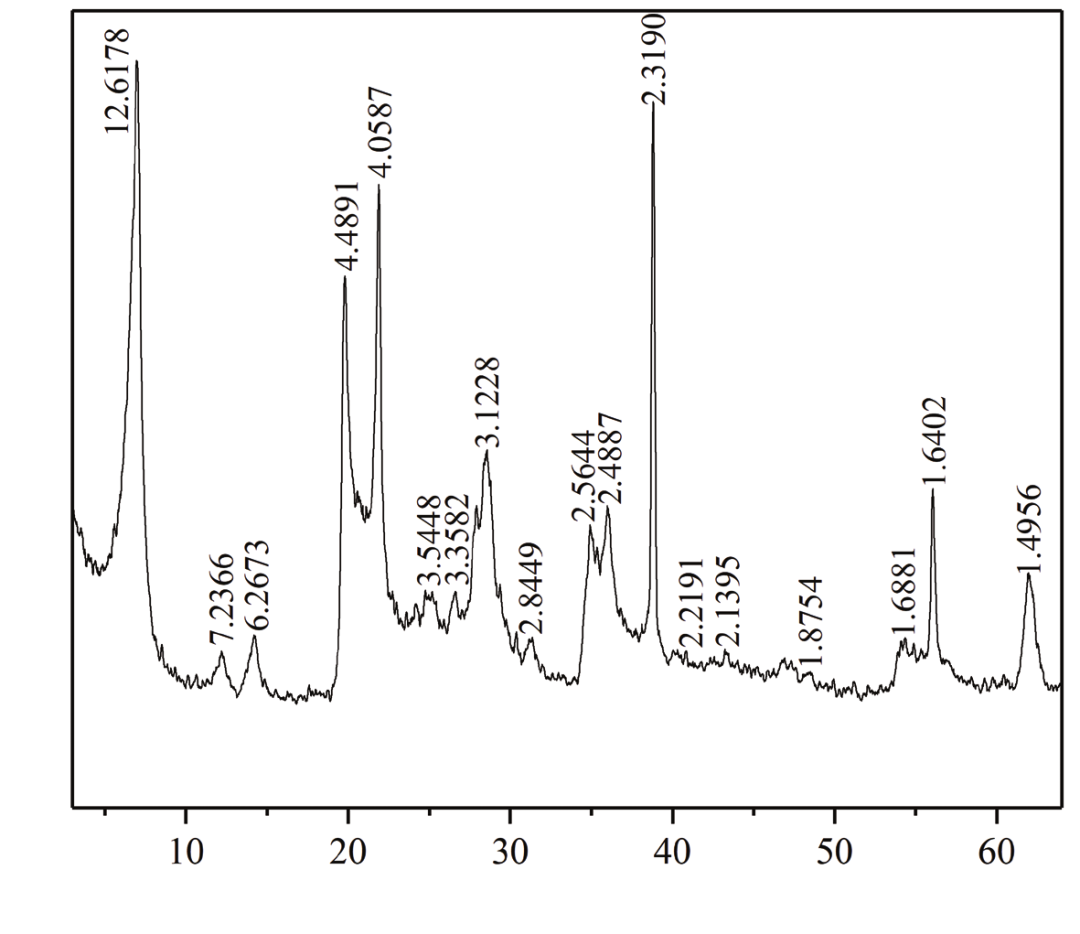
如果样品为非晶质,衍射背底较高。
含非晶质蒙脱石谱图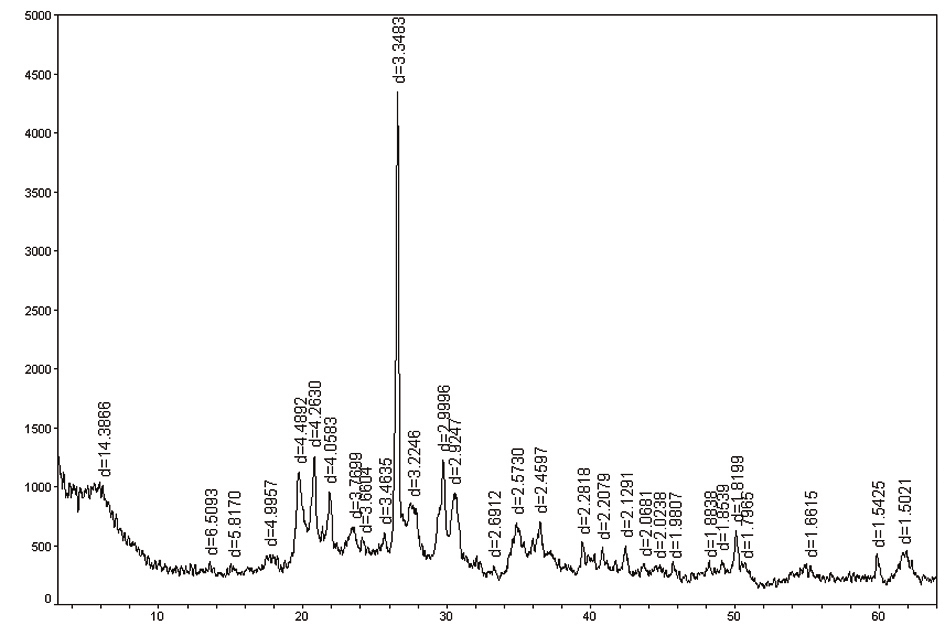
结构分析 煤系针状焦的宏观性能与其微观结构研究
X 射线衍射技术是最常用的重要手段之一,可从纳米尺度研究针状焦的微观结构特征。晶体材料的经典 Bragg-Scherrer 方法不合适,因此有研究者采用分峰拟合方法将结晶碳的衍射峰从体系中分离出来,再通过 Bragg-Scherrer 方法计算样品的微晶结构参数。 范青洁等人采用样品的 XRD 数据,利用 CarbX 软件对其全谱拟合,定量出煤系针状焦原料 SCTP 在不同炭化温度下的微晶结构参数,再与炭化温度关联,进而研究了 SCTP 在成焦过程中微观结构的变化情况,探究机理。
SCTP 及其热处理到 1400℃ 样品的 XRD 图谱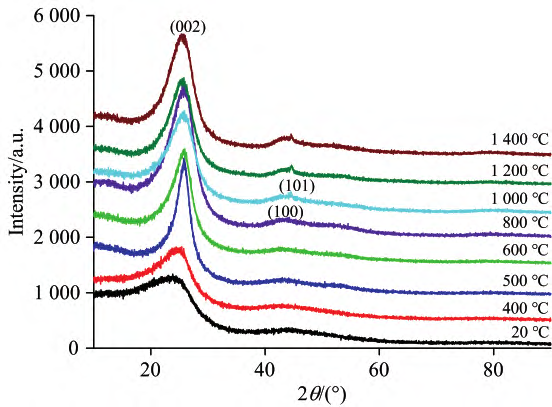
SCTP 及其热处理到 1400℃ 样品的 XRD 图谱的 CarbX 拟合结果(a,b,c)和 SCTP-1400℃ 的分峰拟合结果(d)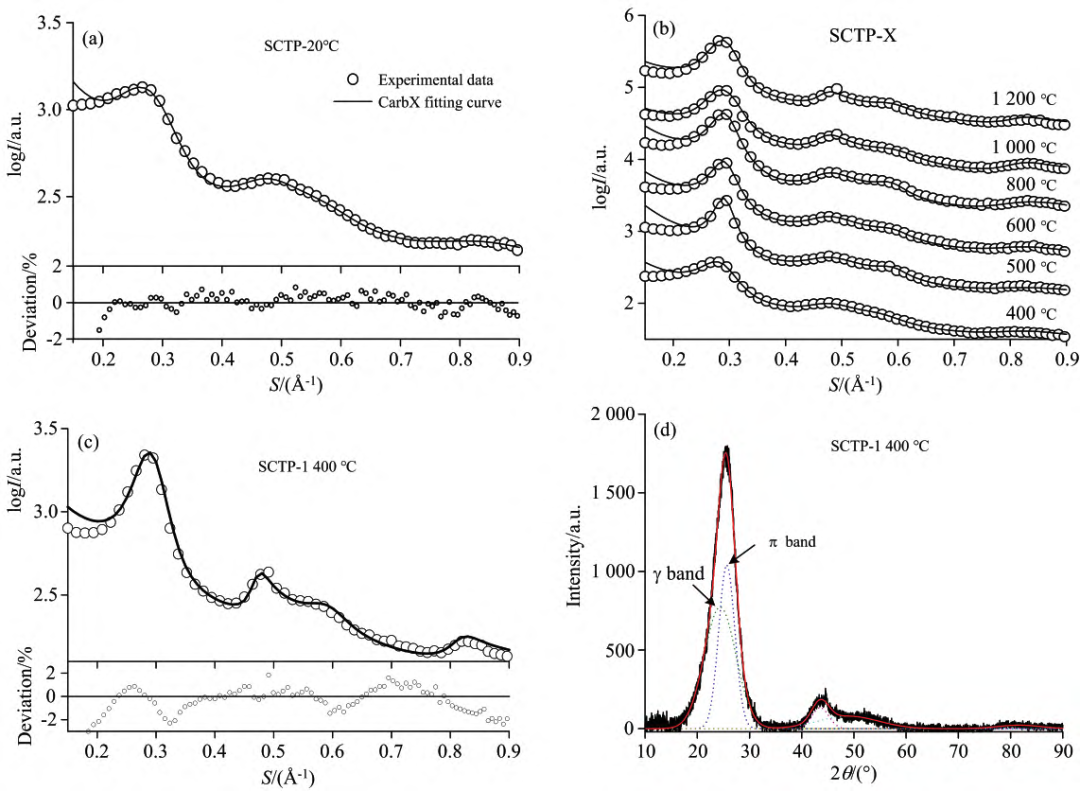
X 射线衍射(XRD)数据处理
XRD 数据采用 MDI JADE 软件进行分析(在分析前,确保自己电脑中同步装有 PDF 数据库)。物相鉴定作为 XRD 最广泛的应用,主要是根据所测得样品衍射图谱与给定检索条件后 PDF 卡片库中的“标准卡片”进行对照,然后根据其三强峰峰位、峰强及样品中的元素进行判定是否存在这种物相。 导入文件 XRD 测试完成一般会导出三种格式文件,我们直接将 .raw 格式文件拖入即可。
无条件检索
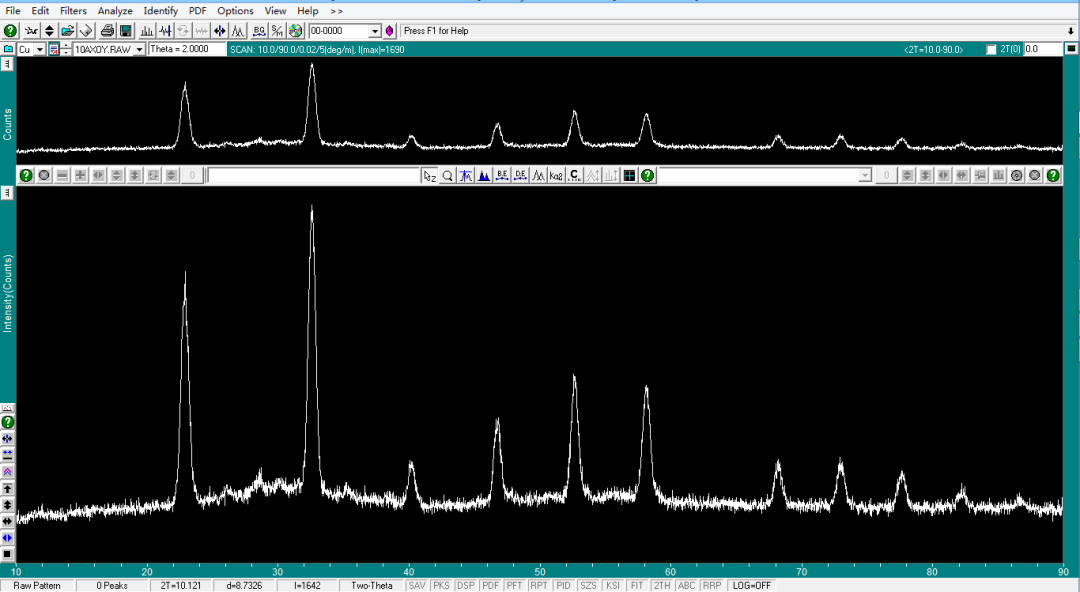
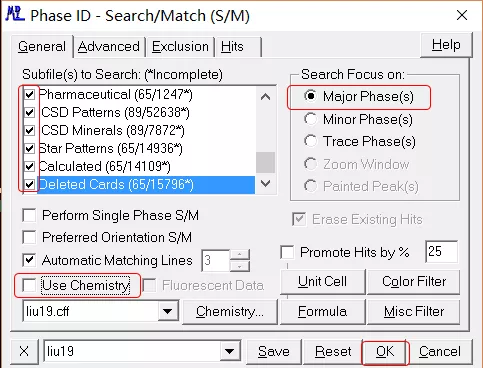
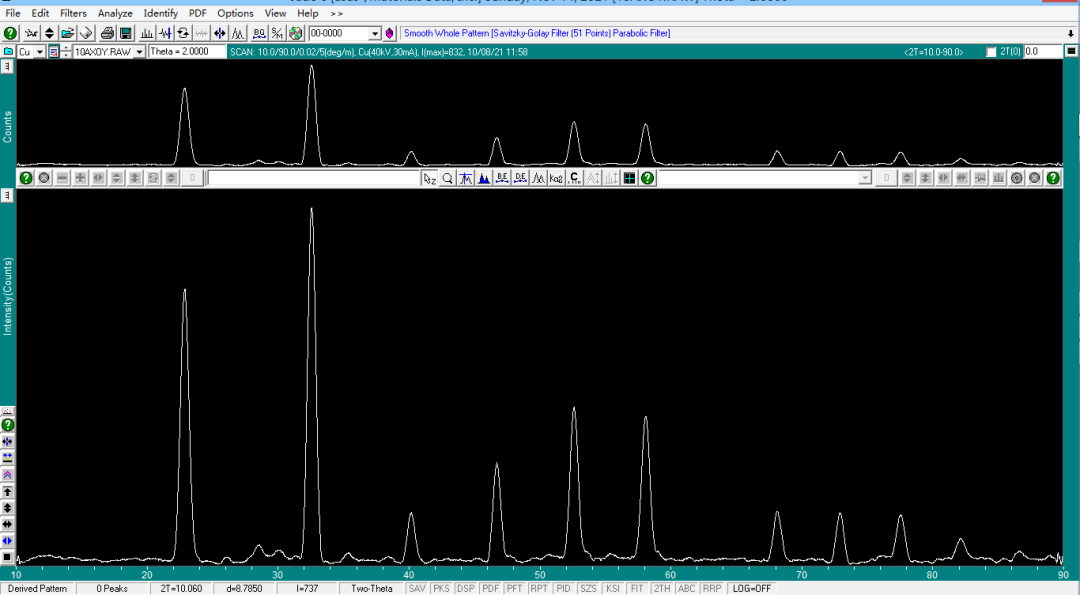
弹出 Search/Match Display 窗口。
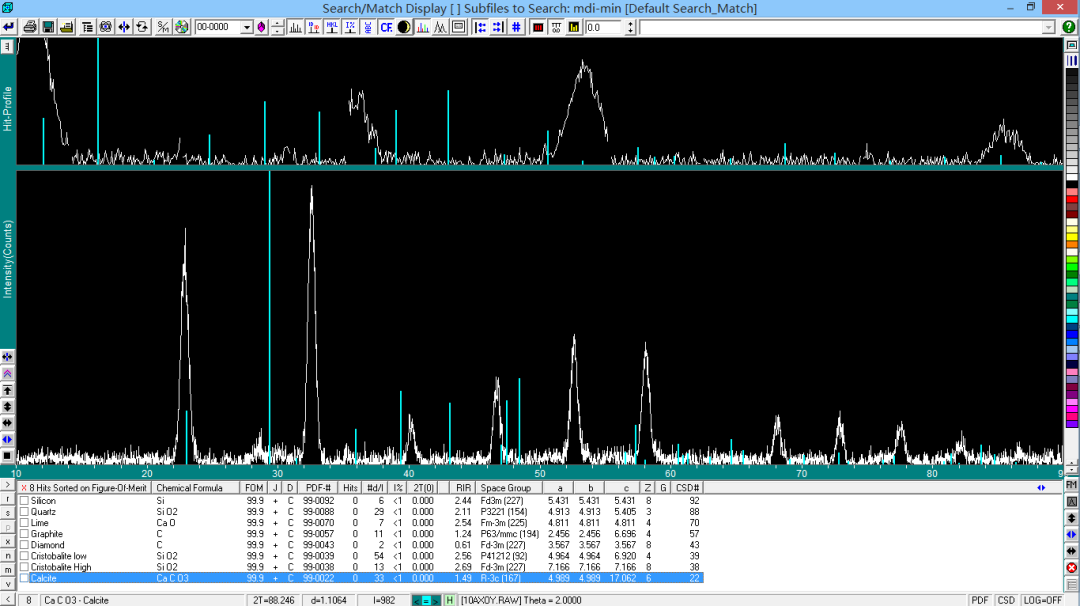
此时,通过点击窗口下列的检索列表可观察 PDF 卡片和衍射谱匹配情况,一般按照 FOM 的值进行排序,FOM 值越小,表示匹配度越高。
限定元素检索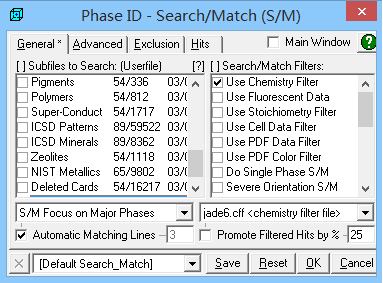
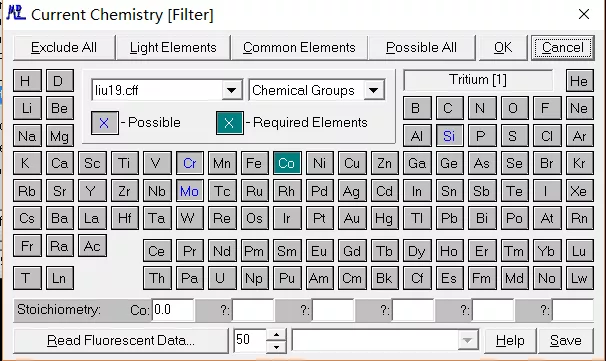
选定元素之后,点击右上角 OK 键,就可以顺利进入曲线拟合阶段。此时,我们可以看到峰位的匹配度已经很高了。
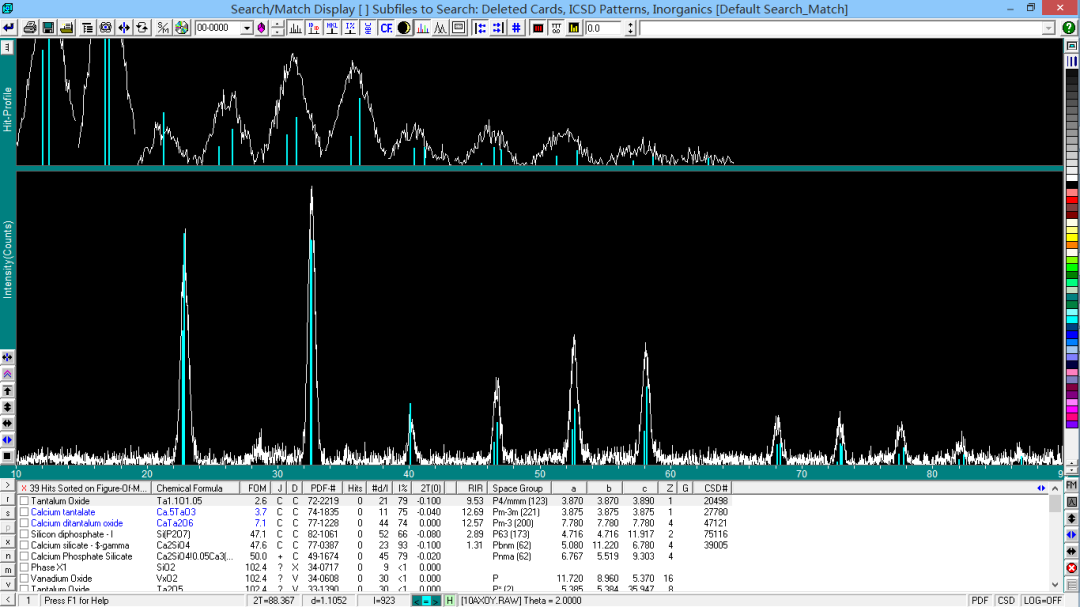
对于一般的样品,进行上两轮检索基本可以确定样品所含物相,但如有仍不能检索出来的物相存在,可采用单峰检索来确定物相。 在主窗口中选择“计算峰面积”按钮,在峰下划出一条底线,该峰被指定,鼠标右键点击“S/M”,此时,检索对象变为灰色不可调(Jade 5 中显示为“Painted Peaks”)。 此时,可以限定元素或不限定元素,软件会列出在此峰位置出现衍射峰的标准卡片列表。
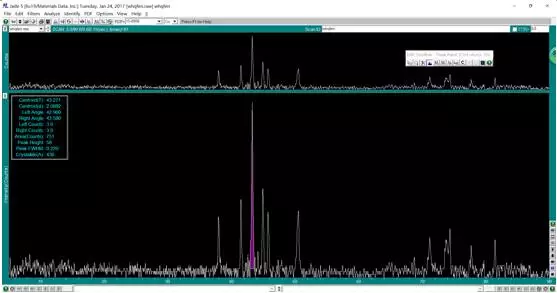
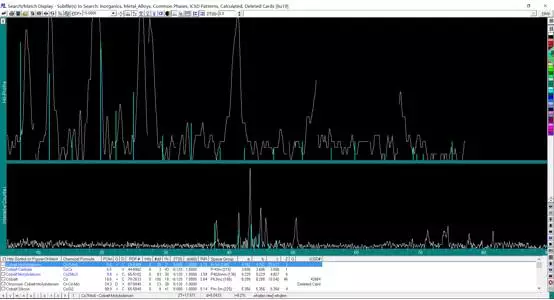
双击搜索出来的物相,弹出相应的 PDF 卡片,从而可以获得对应的晶面指数,2-theta 等信息。
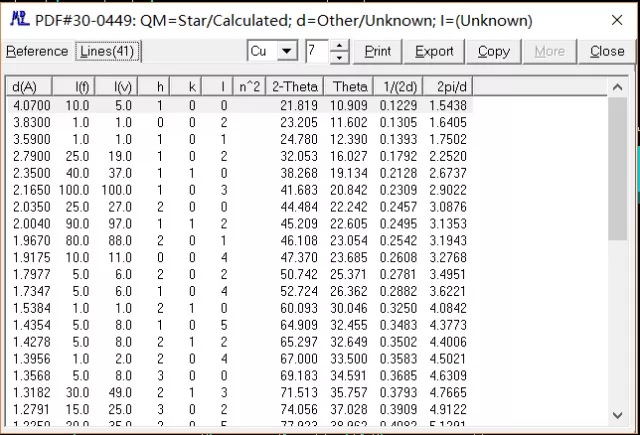
晶粒粒度分析 衍射粉末晶粒大小的计算主要是以衍射图谱的半宽高为依据来进行相关计算。 如果把衍射峰简单地看作是一个三角形,那么峰的面积等于峰高乘以一半高处的宽度。这个半高处的高度有个专门名词,称为“半高宽”,英文写法是 FWHM。 具体的操作过程如下:如上所示进行物相检索,在确定对应物相后,右键单击常用工具栏中图片按钮,设置扣除 Kα2,进行扣除背景操作。图中红框打勾即可。
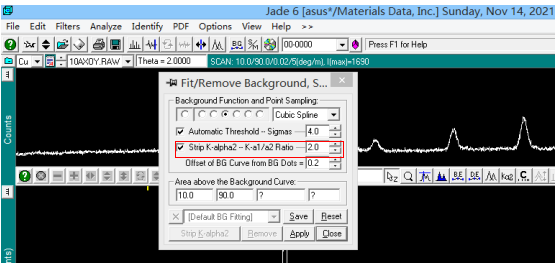
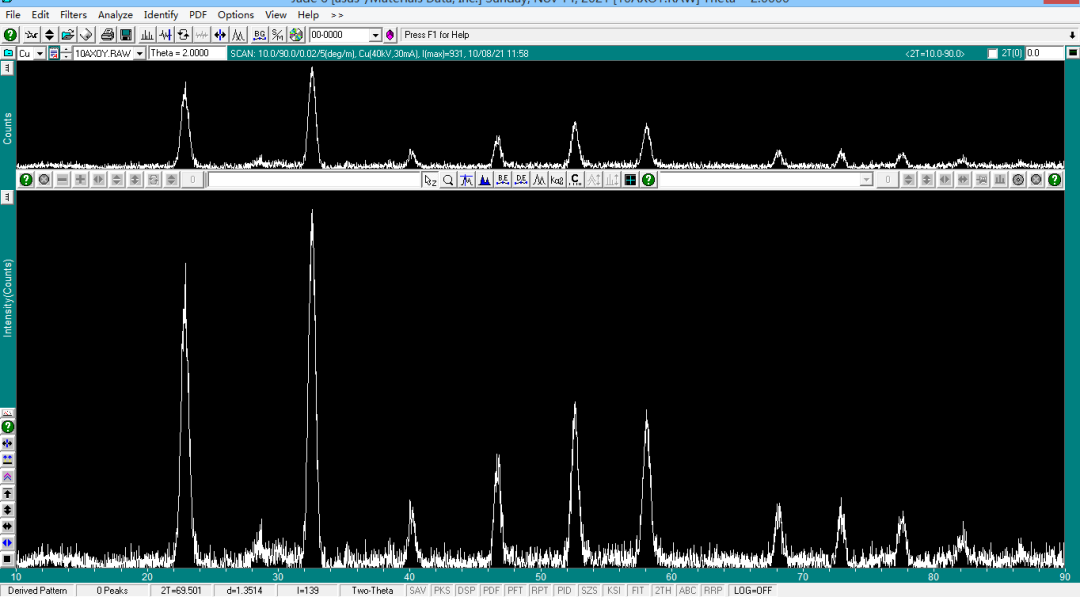
去除前:
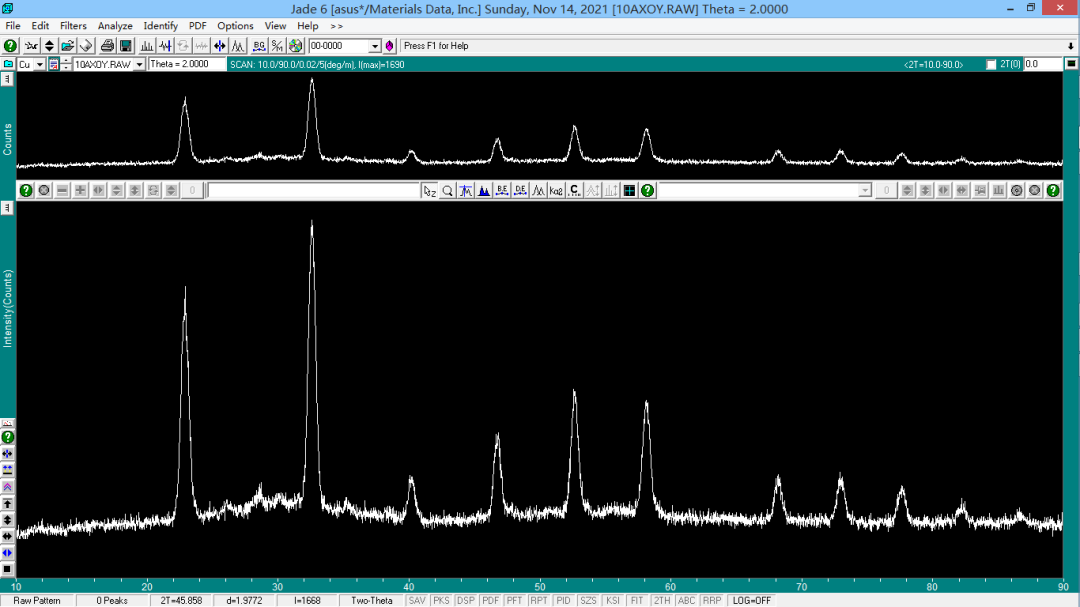
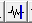
去除后:

注意:利用 XRD 进行晶粒大小计算时,前提是假定晶粒为“球形”,所以其测出来的粒径不是很可靠,结果总是小于 SEM 和 TEM,但无法进行 SEM 和 TEM 时,其结果具有一定的参考依据。 数据作图 目前,大部分科技论文中基本均采用 Origin 软件进行作图处理。作图时,以 2θ 为横坐标,所得衍射强度为纵坐标。具体作图过程此处不采用详细介绍。
X 射线衍射(XRD)常见问题
Q1 为什么 XRD 数据的峰强度较低,甚至没有明显的衍射峰? 样品的衍射峰强度最主要跟样品本身的结晶度有关,其次跟样品量以及仪器的功率都有关系。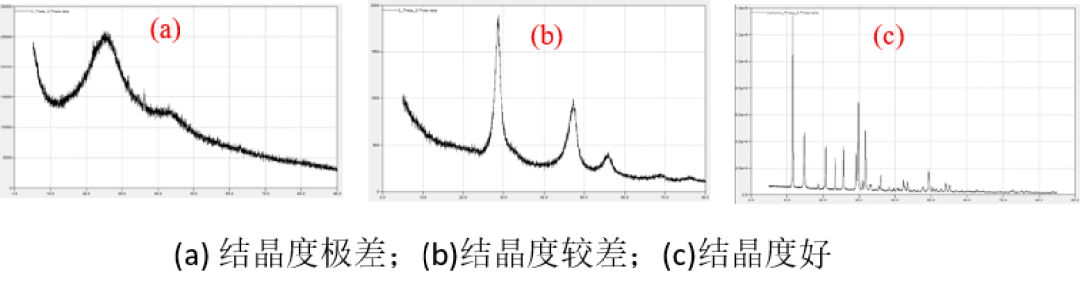
Q2 除了结构缺陷和应力等因素外,为什么粒径越小,衍射峰越宽? 从衍射理论知道,衍射极大和第一极小之间的角宽度与发生相干衍射区域(相干域)的尺寸有关,相干域越大,角宽度就越小。一般来说,相干域的尺寸小于 2 微米,就会使衍射峰造成可测量的宽化。所以,晶粒的粒径越小,以至不能再近似看成具有无限多晶面的理想晶体,对 X 射线的弥散现象就越严重,表现在峰强变弱,峰变宽。
Q3 XRD 衍射强度和峰的宽度与样品颗粒大小,还是与晶体颗粒大小有关? 样品中晶粒越小,衍射峰的峰高强度越来越低,但是峰越来越宽,实际上利用 X 射线衍射峰的宽化对样品的结晶颗粒度分析就是根据这个原理的(Scherrer 公式)。 晶粒大小和颗粒大小有关系,但是其各自的含义是有区别的。一颗晶粒也可能就是一颗颗粒,但是更可能的情况是晶粒抱到一起,二次聚集,成为颗粒。颗粒不是衍射的基本单位,但是微小的颗粒能产生散射。磨的越细,散射就越强。对于晶粒,磨过头了,晶体结构被破坏了,磨成非晶,衍射能力就没有了。磨得太狠的话,有些峰可能要消失了,而且相邻较近的衍射峰会由于宽化而相互叠加,最终会变成一个或几个”鼓包”。一般晶面间距大的峰受晶粒细化的影响会明显一些,因 d 值大的晶面容易被破坏。
Q4 XRD 峰整体向右偏移是什么原因造成的? 可能是离子半径小的元素取代了离子半径大的元素;也可能是制样时,样品表面高出了样品座平面或者仪器的零点不准造成的,建议最好用标样来修正数据。
Q5 影响仪器测量结果的分辨率仅仅取决于 θ 吗? 影响仪器测量结果的分辨率的因素是多方面的:测角仪的半径;X 射线源的焦斑尺寸;光学系统的各种狭缝的尺寸;仪器调整情况;采数步宽;样品定位情况等。
本文源自微信公众号:中科蓝海ZKBO
原文标题:《探索物质微观世界的奥秘:X 射线衍射(XRD)测试(下)》
原文链接:https://mp.weixin.qq.com/s/-TLffy55yf-_R4-iJzJ0DQ
本转载仅出于分享优质测试干货,旨在传递更多观点,并不代表赞同其全部观点或证实其内容的真实性。文章中所包含的图片、音频、视频等素材的版权均归原作者所有。如有侵权请告知删除。