

Q1:朱老师,我确认一下,Cu扩胞后优化需要固定底部原子吗?
A:做吸附需要
Q2:朱老师,在切表面计算slab电子结构的时候出现的表面态要怎么去除呢?
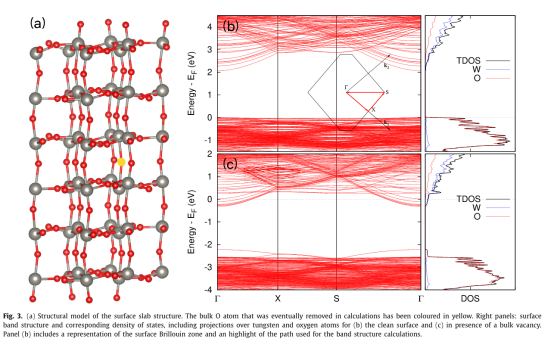

A:末端的w加上o
Q3:朱老师,请问在incar里面添加LDIPOL = .TRUE.IDIPOL = 3 EFIELD = ?问号为外加电势数值,这样可实现恒电势计算吗?
A:恒定电势势给催化剂提供电子调控费米能级
Q4:朱老师,计算co2在催化剂表面吸附弯曲,只转移部分电子是不是不能用计算氢电极,只能用aimd计算?
A:二者没有直接关联。
Q5:朱老师,如果要在阴极添加负电势,调整费米能级是通过更改NELECT标签增加价电子数目实现的吗?增加电子后如果用显示溶剂化,是否需要增加质子保证整体电中性,另外能否给出更改费米能级的具体方法?
A: 通过nelect加电子来计算功函数,调节费米能级,不需要增加质子
Q6:朱老师,双原子催化剂,比如Ni,Zn,设置INCAR时要加MAGMOM吗,如果要加怎么设置呢
A:可以把MAGMOM可以设为5来优化
Q7:朱老师,单原子催化剂如果算出来的吸附能偏大,怎么调,能通过改以下INCAR?还是调结构?
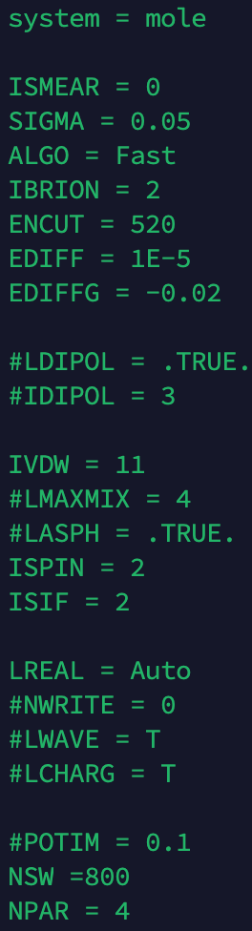
A:调整下吸附分子的吸附构型,比如角度等
Q8:朱老师,在调用vtst的时候出现这个报错怎么办呢? 计算停止了
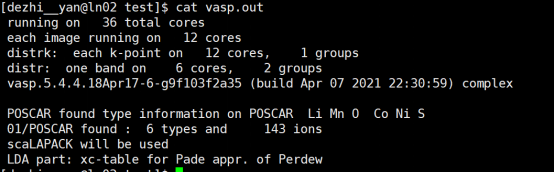
A:这个不是错误信息, 是正常输出,计算停止可能是因为内存不足
Q9:朱老师,我想问一下,假设我要算Li3Al2的电压平台,该用什么公式去算呢,Li3Al2前一个相是LiAl?
A:这个应该是逐步脱出li,算形成能
Q10:朱老师,这里*NH3→*+NH3 脱附最后一步骤台阶图的为啥有些人计算都不一样呢,最后一步(*+NH3)是在同一个体系里面定值吗?
A:应该是确定值,因为和催化剂无关了,热力学表上有
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!