

Q1:朱老师,结构优化成功收敛,但是在结构自洽的时候不收敛要添加什么参数呢?
A:可以读取波函数,ISTART=1
Q2:朱老师,我的功函数的费米能级算的太低了,做出来的图不好看,要怎么调整呢?
A:减小真空层厚度
Q3:朱老师,计算完一步以后就报这个错误是什么原因呀?
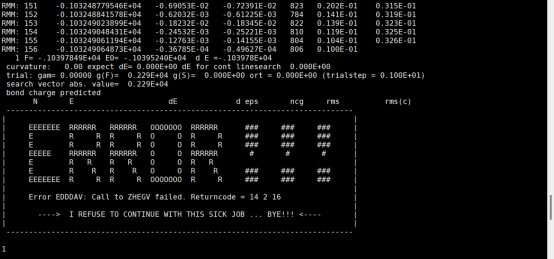

A:换下ibrion试试
Q4:朱老师,我在吸附NO的时候是以end形式,但是到NHO的时候就变成类似side-on的,这种路径还要考虑吗?
A:可以考虑,变形很正常
Q5:朱老师,我想问一下,在VASP计算中,用过渡金属(用TM1表示)对过渡金属(用TM2表示)氧化物掺杂的时候,我知道TM2需要加U;但是TM1,也就是过渡金属掺杂物,这个也是否需要加U呢?谢谢
A:都要加
Q6:朱老师,如果用PW91方法,计算时除了POTCAR用PW91的POTCAR,以及GGA=91,别的地方还有和使用PBE不同的地方吗
A:其他不用改了
Q7:朱老师,请问计算NO小分子时候,ispin要不要打开呢?
A:可以不开
Q8:朱老师,我在用PW91方法跑Cu原胞优化的时候会有这种报错,是什么地方不对呢?
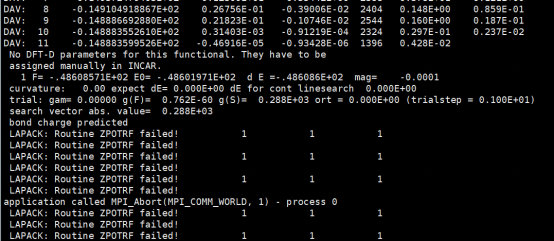
A:把IVDW关掉
Q9:朱老师,DOS计算的时候K点的增加是在自洽计算就加上还是计算dos的时候加上?
A:前面的自洽计算就加上,DOS时直接读取波函数和电荷密度,后面再加,可能会报错。
Q10:朱老师,催化剂是Cu(111)加上CF2一起作为催化剂再吸附别的分子,那我在扩胞之后,优化催化剂的话是优化有CF2的?还是先Cu(111)优化,再加上CF2再优化一次,分两步呢
A:分两步吧,Cu单独优化一下
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!