

Q1:朱老师,计算FE110表面H2的吸附时,需要考虑色散校正开启IVDW=12吗?
A:需要开启,H₂在 Fe (110) 表面的吸附通常属于化学吸附,而非纯粹物理吸附;
研究分子吸附,H₂以完整分子形式存在,如低温预吸附:必须开启IVDW=12
解离吸附(主要场景):推荐开启IVDW=12,尤其当需要定量结果时,误差更小,与实验一致性更好;
分子吸附(次要场景):必须开启IVDW=12,否则会严重低估吸附能。
Q2:朱老师,异质结计算时算法选择有什么规则吗?
A:不同算法(DFT-D2、DFT-D3、DFT-TS 等)的选取需结合异质结的相互作用强度、层间距离和计算精度需求;
异质结的层间相互作用可分为弱范德华主导 和 强耦合两类,算法选取需匹配这种特性;
DFT-D3(IVDW=11/12):推荐 “多数范德华异质结”(优先选 D3 (BJ))。
Q3:朱老师,rmm算法会停在12卡住,dav算法是跳出error:EDDDAV?
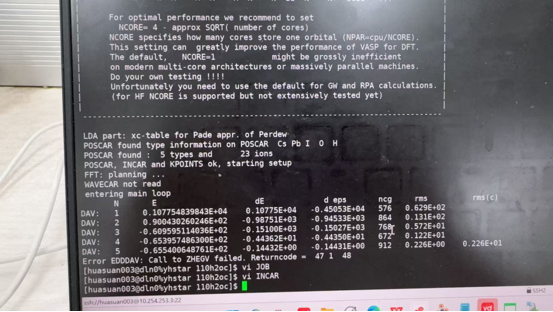

A:调节一下amix试试,0-0.4之间;
水分子与表面的 初始结构不合理,引发强排斥或电荷振荡;
可能是算法特性与体系不匹配;
或者收敛参数设置过严或迭代步数不足。
Q4:朱老师,计算弹性常数时总的自由度直接从33增加到了126,计算量增大好多?
A:缩小晶胞,否则就是那么多;
减小超胞或调整掺杂浓度降低原子总数;
弹性常数的独立分量由晶系对称性决定(如立方晶系仅需 3 个独立常数:C₁₁、C₁₂、C₄₄),无需计算所有可能的应变模式,可大幅减少总计算量。
【做计算 找华算】
华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。