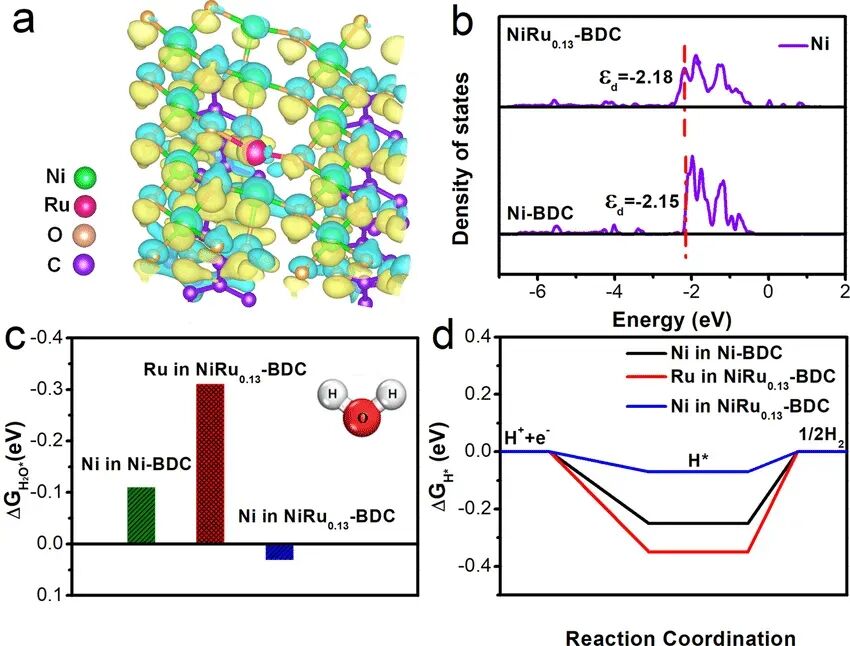
VASP(Vienna Ab initio Simulation Package)是一款基于密度泛函理论(DFT)的量子力学模拟软件,广泛应用于材料科学、化学、物理学等领域。其核心计算方法之一是自洽场(Self-Consistent Field, SCF)算法,解决Kohn-Sham方程,该算法通过迭代计算电子波函数和密度,直到系统达到自洽状态。
自洽计算是VASP计算中最基础也是最重要的步骤,其目的是找到电子波函数和相应的总能量,使得电子在晶胞中的分布达到自洽。自洽计算通常包括电荷密度的初始化、波函数的迭代优化以及能量和力的计算。
VASP计算需要准备超算连接软件EASYCONNECT与SSH,建模软件VESTA,超算连接软件Winscp
jp-minerals.org/vesta/en/download.html
EasyConnect下载-EasyConnect最新版下载V7.6.7.0
Downloading WinSCP-6.5.3-Setup.exe :: WinSCP
cp CONTCAR POSCAR 把优化的结构保留下来
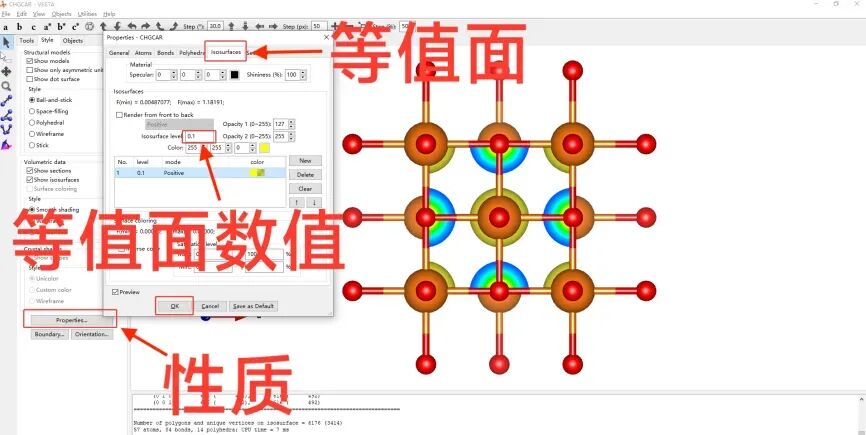
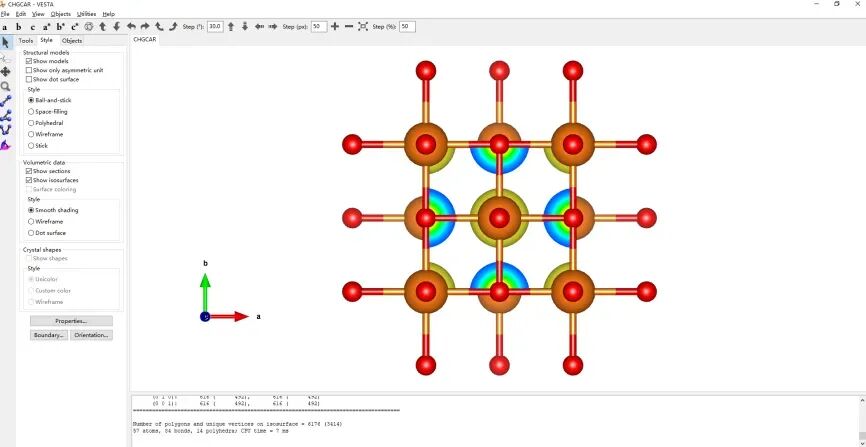
vi INCAR 加入自洽计算和Bader电荷标签,修改标签IBRION=-1、NSW=0,增加LAECHG=.T.
chgsum.pl AECCAR0 AECCAR2 价电子与芯电子求和
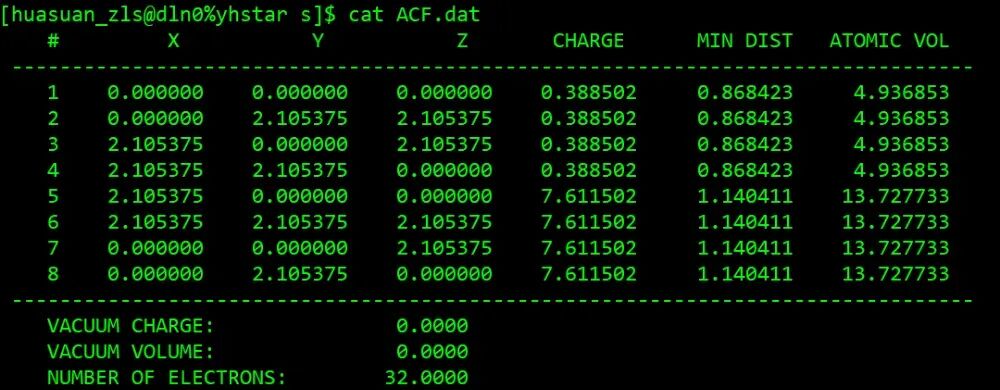
bader CHGCAR -ref CHGCAR_sum
通过本文的学习,大家已经能够数量掌握VASP电荷密度计算功能,包括自洽计算设置、电荷密度分析、Bader电荷分析。
在此基础上,你更形成了 “计算 – 分析 – 解读” 的完整能力闭环 —— 这些技能将为你深入探究材料电子结构本质、解析化学键成键特性、量化元素间电荷转移规律等关键科研问题提供坚实支撑,助力你的研究工作从定性描述迈向定量分析的精准维度,为催化机理阐明、功能材料设计、新型体系开发等方向的突破奠定核心基础。
本页内容为VASP零基础系统化教程的单个章节。为了让您的学习更加高效、体系化,避免碎片化知识的困扰,建立坚实的知识框架,我们诚邀您访问 【VASP零基础系统化教程合集】 页面。该合集整合了全部教程,形成了一套严谨的 “理论奠基 → 工具准备 → 参数实操 → 场景实战 → 结果分析” 四阶一闭环学习体系。遵循此路径,您将能循序渐进地掌握VASP计算模拟的核心技能。
? 访问合集,获取完整教学体系 >>
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!