

Q1:朱老师,我在计算钙钛矿构型的LaCoO3时,Co和O构成八面体结构,但是Co的eg和t2g轨道并没有按照八面体模型那样分裂,而是混在一起,请问可能是什么问题呢
A:完全分裂是理想化的结构,在固体中电子不在是分立的能级,而是能带,肯定会有交叠
Q2:朱老师,是不是简并态的话用能带而不是态密度看比较合适?
A:其实态密度,能带都是一样
Q3:朱老师,算这种扩散的时候,锂原子要距离基底多远比较好?
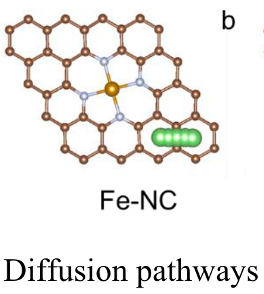

A:两个埃左右
Q4:朱老师,算这种分子扩散能垒,起点终点过渡态,三个点就行了是吧?起点和终点位移距离2~3埃可以吗?
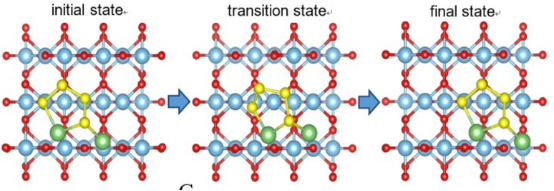
A:这么多原子迁移可能不太好算,可能不收敛
Q5:朱老师,请教个LiS建模的问题,针对Li2S4 ~ Li2S8模型,文献中的优化后的结构完全不一样,看不出所以然。所以,不太明白这个在建模中,Li-S,键长和角度有什么要求吗?相对于Li2S和Li2S2,Li2S4 ~ Li2S8模型具有更多的原子,空间构型是否也要考虑?
A:从s8基础上修改,再结构优化
Q6:朱老师,Li2S1 ~ Li2S8是否具有固定的结构,就像水分子一样?如果是的话,其键长,键角应该是固定的
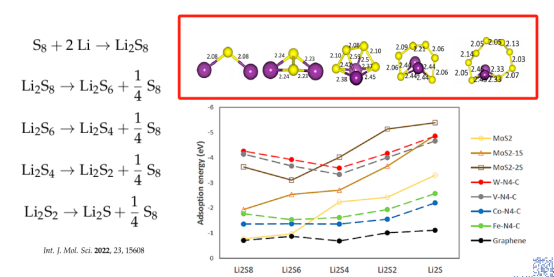
A:照上面建立结构模型,然后吸附到催化剂表面
Q7:朱老师,能垒路径有些值是负数正常吗,算能垒应该哪个值减哪个值?
A:一般头尾比中间要低,中间有负值的就不正常,要重新算
Q8:朱老师,我想问一下,如果我计算单层MoS2对多硫化物的吸附作用,那么我应该怎么固定原子呢?一共就三层原子
A:不需要固定
Q9:朱老师,请问异质结的距离一般设置为多少合适呢?
A:三个埃左右
Q10:朱老师,我发现结构优化完后,Li-S结构经常散了,这种情况,怎么处理呢?
A:这个只能再调整初始构型了
本次答疑由拥有15年VASP实战经验的华算科技朱老师(同济大学本博、深圳海外高层次人才)提供。?点击进入《VASP | 理论计算常见问题解答专题》,快速查找更多计算解决方案。