

Q1:朱老师,请问过渡态计算过程中的精算是在粗算之后,将每个文件夹的CONTCAR复制为POSCAR,修改精度和算法后接着计算嘛?
A:是的
Q2:朱老师,计算过渡态的时候出现这个情况是什么原因呢,初态和末态位置差距很大?这个末态一优化就这个样子了
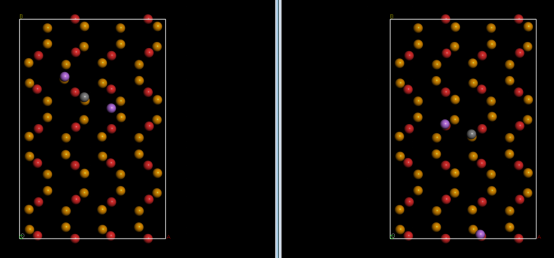

A:散架了,换个算法或者路径试试,改改初始结构。
Q3:朱老师,算隐式溶剂化效应结构老散架怎么办?
A:散架和溶剂效应关键不大,检查一下原始结构
Q4:朱老师,请问nebef.pl这个脚本第二列的能量,为什么与结构优化后的那个能量有出入呢?我初态结构优化能量是-220.250,但是用nebef.pl的时候我的初态能量就显示为-220.244200?
A:f和e0的差别吧,脚本读取的是e0
Q5:朱老师,请教一下 在计算能带时候报错 这是什么原因呢?
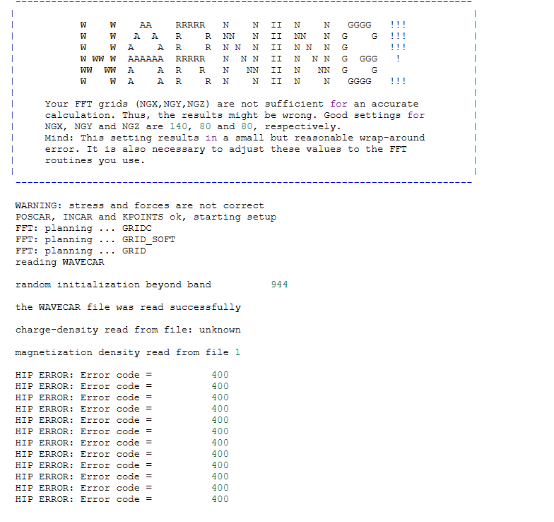
A:不读取波函数试试,把电荷也去了试试
Q6:朱老师,我用vaspsol计算隐式溶剂效应,但是算出来的能量和真空中自洽的能量几乎一样?
A:一般在自洽计算加这个,数值接近是正常的,0.1。
Q7:朱老师,假如单个金属原子在催化剂表面的结合能很大,且大于多个金属原子团聚在一起的内聚能,能不能说明这个金属原子有在催化剂中形成单原子的可能?
A:可以的,更加稳定
Q8:朱老师,我在计算功函数时报错 请问如何解决呢?
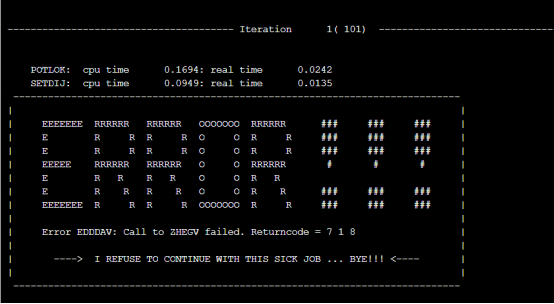
A:设置一下amix=0.1试试
Q9:朱老师,金属原子吸附H,电子步很难收敛有什么解决办法吗,能量反复震荡,1e-1到1e-4之间,120个电子步也不收敛,一系列类似的结构都是如此,ALGO=N,自旋极化体系,调整过AMIX也没什么效果,ENCUT是600eV,大约是ENMAX的1.5倍,离子步也很难收敛,算了几十步报错如下
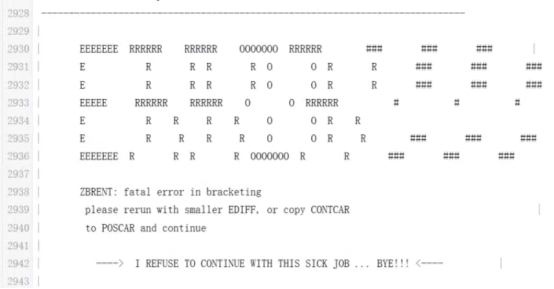
A:先降低截断能试试
Q10:朱老师,自洽计算提示这个,此时收敛的结果可信吗?并没有设置NELECT?

A:设置amix试试,调整数值
本次答疑由拥有15年VASP实战经验的华算科技朱老师(同济大学本博、深圳海外高层次人才)提供。?点击进入《VASP | 理论计算常见问题解答专题》,快速查找更多计算解决方案。