VASP(Vienna Ab initio Simulation Package)是一款广泛应用于材料科学和计算化学领域的第一性原理量子力学软件,能够进行从原子尺度到分子尺度的模拟和计算。
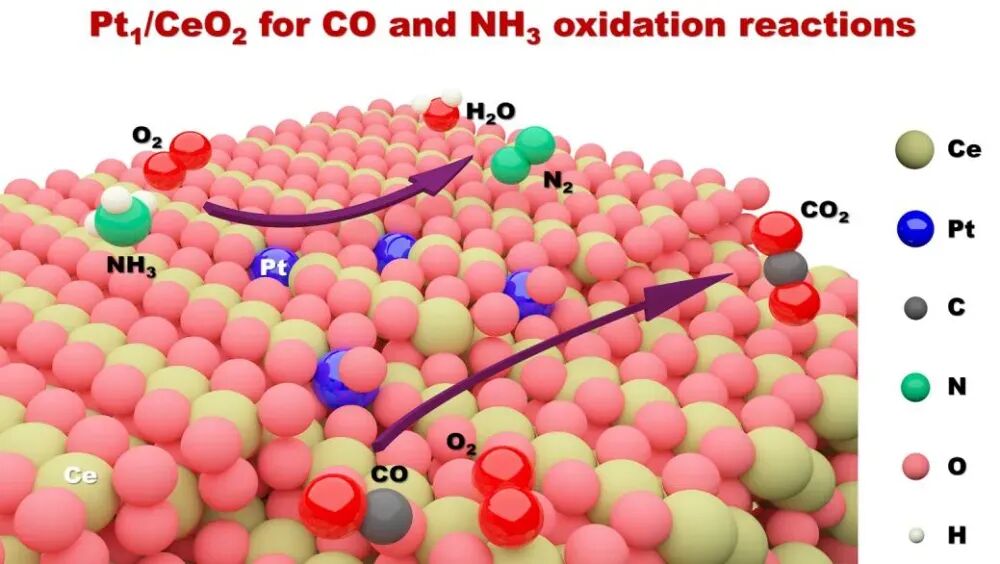
在材料科学中,表面能是描述固体物质表面稳定性的关键参数,对于理解材料的催化性能、吸附行为、界面性质等具有重要意义。
华算科技朱老师将详细探讨VASP在表面能计算中的高级技巧,涵盖计算步骤、参数设置、常见问题及优化方法,以帮助用户更高效、准确地进行表面能计算。
表面能是描述固体表面能量状态的物理量,其定义为单位面积表面相对于体相的多余能量。在VASP中,表面能的计算通常通过构建表面模型(slab模型),并进行结构优化和能量计算来实现。表面能的计算公式通常为:
其中,Erel 是弛豫后的表面能量,σunrel 是切开体相的功,Natoms 是表面原子数,A 是表面积。
在VASP中,表面模型的构建是表面能计算的第一步。构建表面模型时,需要考虑以下几点:
选择合适的晶面和超胞结构,以确保模型的周期性和对称性。例如,对于Ni表面,可以选择(100)、(111)等晶面进行建模。
在表面模型中添加足够的真空层,以防止不同表面单元之间的相互作用。真空层的厚度通常根据体系的性质和计算精度要求进行调整。
在构建表面模型后,需要进行结构优化,以达到能量最低的稳定结构。结构优化通常包括晶格参数优化和原子位置优化。在VASP中,可以通过设置ISIF参数控制结构优化的范围,例如ISIF=2表示同时优化原子位置。
在VASP中,计算表面能时,参数的设置对计算结果的准确性和效率至关重要。以下是一些关键参数的设置建议:
平面波截断能决定了计算的精度。通常,ENCUT值越大,计算精度越高,但计算时间也越长。建议根据体系的性质选择合适的ENCUT值,例如ENCUT=400或ENCUT=500。
ISMEAR参数控制电子占据态的模糊化方法。常用的ISMEAR值包括ISMEAR=0(Gaussian方法)和ISMEAR=1(Methfessel-Paxton方法)。选择合适的ISMEAR值可以提高计算的稳定性和收敛性。
K点网格的设置对计算的精度和效率有重要影响。通常采用Monkhorst-Pack方案生成K点网格,以确保计算的收敛性。
EDIFF和EDIFFG参数控制能量和力的收敛标准。建议设置适当的收敛标准,以确保计算的稳定性和准确性。
在VASP中进行表面能计算时,一些高级技巧可以提高计算的效率和准确性:
在几何优化过程中,可以适当放宽收敛限制,例如将EDIFFG设置为-0.03或-0.05,以提高计算效率。
对于复杂体系,可以采用多尺度计算方法,例如结合第一性原理计算和分子动力学模拟,以提高计算的准确性和效率。
对于大规模计算,可以利用并行计算和资源优化方法,例如使用高性能计算集群和优化计算资源分配,以提高计算效率。
在VASP中进行表面能计算时,可能会遇到一些常见问题,例如:
如果结构优化失败,可能是由于初始结构不合理或参数设置不当。建议检查初始结构和参数设置,确保计算的稳定性和收敛性。
如果计算结果不收敛,可能是由于收敛标准设置不当或参数设置不合理。建议调整收敛标准和参数设置,以提高计算的稳定性和准确性。
如果计算时间过长,可能是由于计算规模过大或参数设置不合理。建议优化计算参数和资源分配,以提高计算效率。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!