

说明:离子液体是由阴、阳离子在室温或近室温条件下构成的液态盐。由于缺乏传统溶剂的屏蔽作用,其内部存在强烈的静电相互作用和显著的离子–离子关联效应。
为深入理解此类结构–性能关系,研究通常采用以下方法:利用密度泛函理论(DFT)解析电子结构与化学相互作用;通过经典分子动力学(MD)模拟研究热力学与动力学行为;借助QM/MM方法处理局部化学反应;采用粗粒化模拟拓展至更长时空尺度;并结合机器学习技术实现快速性能预测与逆向设计。
离子液体的定义


离子液体是一种完全由离子构成的熔融盐。以我们日常生活中熟悉的食盐(氯化钠)为例,氯化钠需要加热到惊人的800℃以上才能熔化成液体,而离子液体的熔点则低于100℃,许多甚至在零下几十度仍能保持液态。
其背后的奥秘在于离子的结构设计。离子液体则通常由体积庞大、结构不对称的有机阳离子(如咪唑、吡啶、季铵、季鏻离子等)和电荷分散的有机或无机阴离子(如六氟磷酸、四氟硼酸、三氟甲磺酸离子等)组成。
DOI:10.3390/ijms21207745
这种独特的结构带来了几个革命性的特性:首先,大的离子尺寸和低对称性使得离子难以整齐排列形成晶体,从而显著降低了熔点。
其次,阴阳离子间的库仑力极强,导致其蒸汽压极低,几乎不挥发,消除了传统有机溶剂挥发性带来的易燃、易爆、空气污染和健康危害等问题。
再者,其液态温度范围极宽,可达300-400℃,提供了广阔的操作窗口。最后,通过改变阴阳离子的种类和组合,可以对其物理化学性质(如极性、溶解度、酸碱性、粘度、电导率等)进行“量身定制”,以实现特定的功能。这种可设计性是离子液体最特殊的特质。
最后,由于不存在传统溶剂的屏蔽作用,离子液体内部呈现出强烈的静电耦合与多体离子–离子相关,这会显著影响局域结构、动力学与宏观输运性能。
通过多尺度、多保真度的计算策略,研究者可以在无法直接从实验分离出的层次上预测机制并指导离子液体的定向分子设计。
DFT视角解析离子液体
密度泛函理论(DFT)在研究离子液体时主要用于描述分子级的电子结构与化学反应性,是量化离子间局部相互作用、配位、氢键和电子能级(如HOMO/LUMO、带隙)最直接的工具。
常见用途包括计算离子对结合能、溶剂化自由能、氧化还原电位和吸附在电极表面的构型与电荷转移。
实操时要特别注意泛函与色散修正(如DFT-D3或vdW-DF)对能量与几何的影响,杂化泛函在描述电荷局部化与带隙上通常优于GGA但计算成本更高;周期性平面波计算适合研究界面或薄膜,簇模型适合快速评估局部化学反应。
由于DFT本身限制于几十到几百原子的系统规模,常用于为更粗尺度模型提供参考数据与参数(例如力场的电荷分布、二级势能面)。
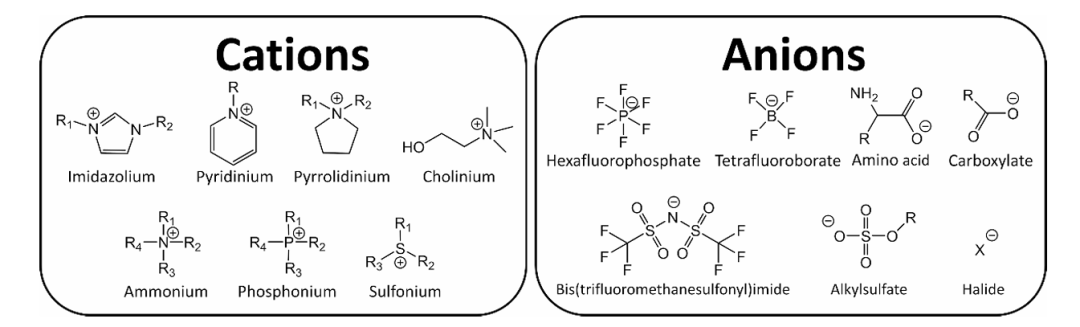
下图展示了使用DFT揭示了1-乙基-3-甲基咪唑阳离子([Emim]+)和 ([BF4]–、[PF6]–、[Cl]–、[Br]–和 [OAc]–) 阴离子构成的离子液体的静电势分布,阴离子中的氧原子及卤素原子为电负性最强部位,能够与咪唑环上的碳氢键、甲基及乙基产生较强的相互作用。
DOI:10.1016/j.molliq.2021.116641
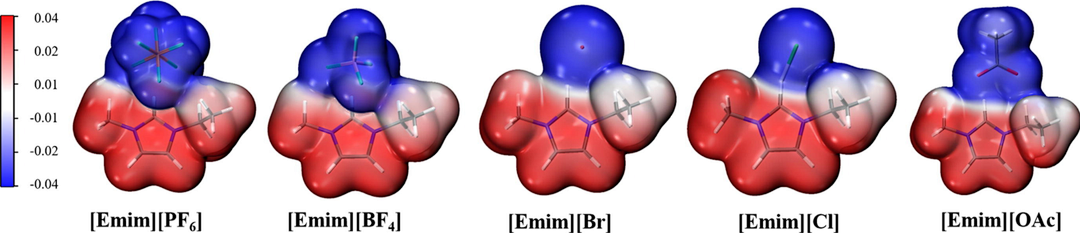
下图展示了1-乙基-3-甲基咪唑阳离子([Emim]+)和([BF4]–、[PF6]–、[Cl]–、[Br]–和[OAc]–) 阴离子构成的离子液体在石墨烯表面的吸附情况显示出明显差异,大小顺序为G-[Emim][PF6]>G-[Emim][BF4]>G-[Emim][Br]>G-[Emim][Cl]>G-[Emim][OAc],这种差异主要归因于阴离子中原子电负性及离子尺寸的不同。
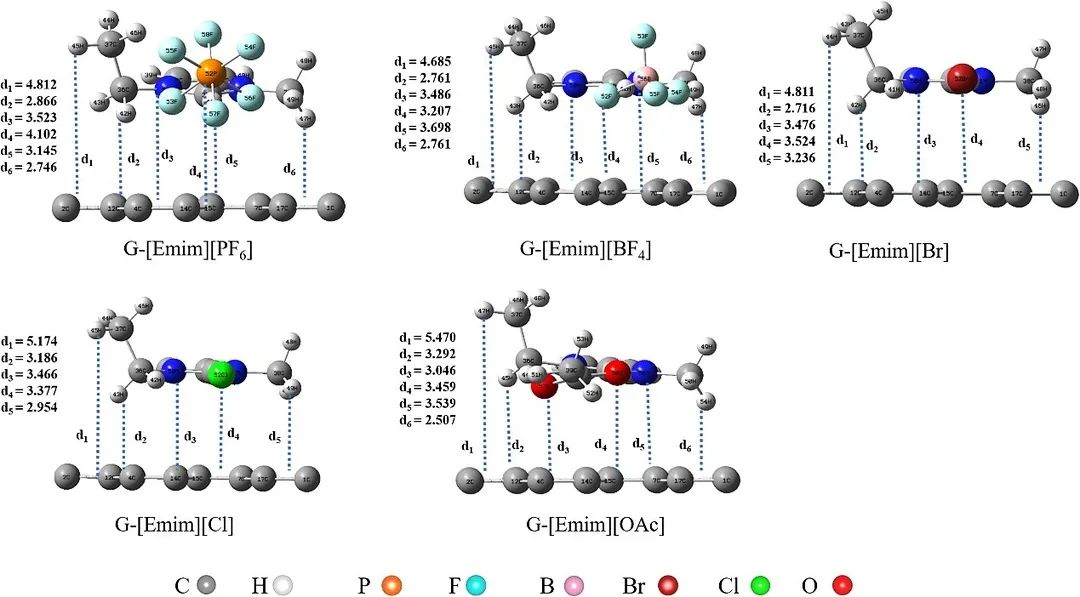
DOI:10.1016/j.molliq.2021.116641
下图则展示了1-乙基-3-甲基咪唑阳离子([Emim]+)和([BF4]–、[PF6]–、[Cl]–、[Br]–和[OAc]–)阴离子构成的离子液体在石墨烯表面的差分电荷密度图,在阳离子下方的石墨烯区域,电子密度增加带负电;而在阴离子下方的石墨烯区域电子密度减少,显示带正电。
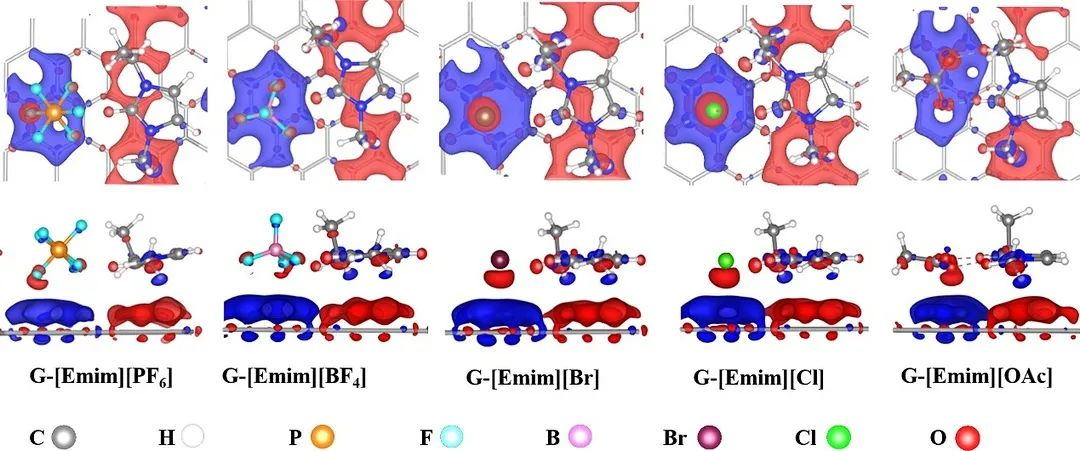
DOI: 10.1016/j.molliq.2021.116641
MD视角解析离子液体
分子动力学(MD)以经典力场描述离子液体的热力学与动力学行为,其关键在于力场的选择与验证:无极化(如CL&P、OPLS/AMBER改参数)能在许多情形下给出合理的结构,但在高电场或强配位场景下会低估极化效应;极化力场(Drude、AMOEBA)更真实但计算费用昂贵。
下图展示了不同离子液体力场在描述双电层的差别,全原子力场的阳离子表现出由单吸附峰到双吸附峰的现象,即“拥挤”(Crowding)现象,此时平行电极排布的离子转为部分垂直电极排布。
而粗粒化力场不存在双吸附峰特征,其离子取向分布几乎不随电势发生变化,始终与电极壁面保持平行(图3f),导致无法捕捉“拥挤”状态。
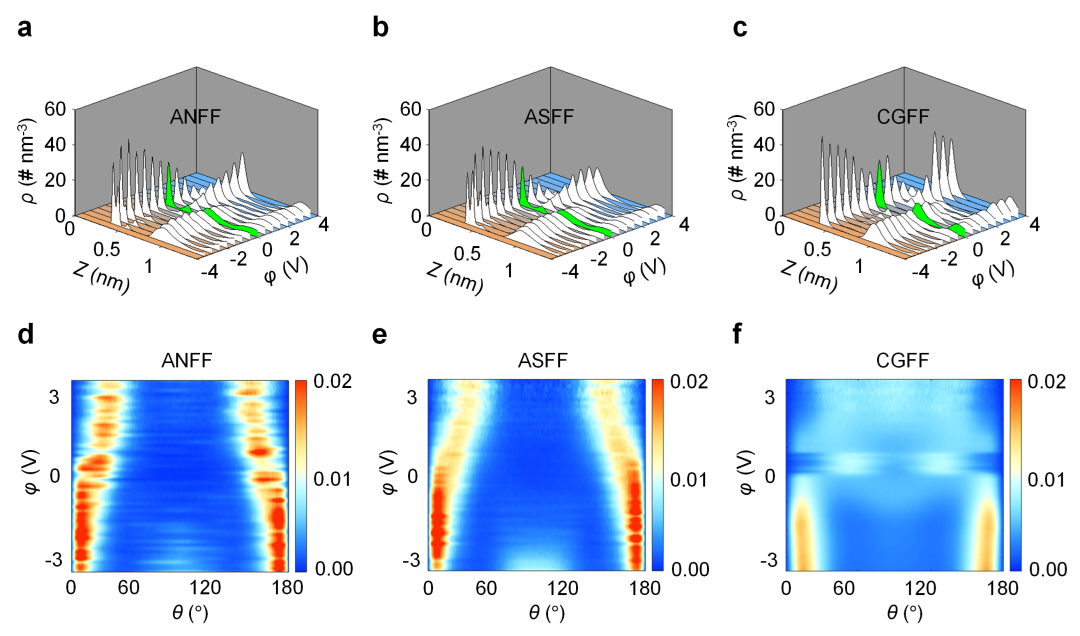
DOI: 10.1039/D4TA00701H
离子液体的长程库仑相互作用需要合适的处理(Ewald/PME),且计算电导率常用Green-Kubo公式或平衡/非平衡方法获得;黏度则依赖长时间尺度的采样(常需数十到数百ns,甚至µs)。
实践建议是用DFT结果校准局部相互作用,用多个体系尺寸和温度点做收敛测试,并结合实验密度、扩散率和谱学数据进行验证。
QM/MM视角解析离子液体
QM/MM把量子力学(QM)区域与经典力学(MM)区域耦合起来,非常适合研究离子液体中涉及化学键重排或电子转移的局域过程,例如电极界面发生的电化学反应、溶剂化诱导的断裂/重组或催化反应机理。
优势是能够在保留化学精度的同时涵盖更大尺度的环境影响;但需要解决的技术细节包括QM/MM边界的处理(link atom或嵌入势)、电荷耦合与极化响应、以及电子密度跨界的稳定性。
在纯离子液体或其与极性溶剂的混合体系中,离子的存在会产生显著的局域电场,诱导邻近离子或分子发生极化。
下图展示了分别介绍在粗粒化模型中考虑极化效应的两种切实可行的方法,一种是基于经典物理图像的Drude方法,另一种则是包含量子化学静电参数的平均场方法。

DOI: 10.1039/D5IM00021A
对于固–液界面,常采用周期性QM在界面活性位点结合MM描述大量液层和电解质离子,以捕获电荷屏蔽与势降的耦合。
实践建议:用DFT(或更高阶方法)对小模型先定位反应路径并计算势垒,再在QM/MM中评估环境调节效应;注意保持QM区足够大以包含关键相互作用并对链接原子进行敏感性测试。
粗粒化视角解析离子液体
如下图所示,粗粒化模拟将若干原子合并为一或几个“粒子”以显著降低自由度,从而可以高效地模拟到微秒–毫秒、纳米–微米尺度,适用于研究离子液体的自组装、相分离、纳孔/毛细输运、在多孔电极或聚合物基体中的分布等宏观/中尺度现象。
DOI:10.1021/jp3008877
粗粒化模型的优点是能直观地看到相分离结构、界面粗糙度和渗流路径,但代价是丢失原子级电子结构信息(无法直接预测化学反应或精细的电荷转移),且参数的可转移性常受限。
粗粒化力场由于准确的动态特性与高效的计算速度,被广泛用于模拟复杂多孔电极,例如CDC , MOF和HsGDY等的电容与充放电过程。
下图展示了粗粒化离子液体在HsGDY不同堆叠结构的充电动力学。结果表明电极孔壁更为粗糙的AB堆叠结构相对于AA堆叠结构,孔内形成了更强的超离子态,导致孔内自由离子比例增加,有利于离子的分离与传输,提升了双电层电容,并降低了离子的传输电阻。

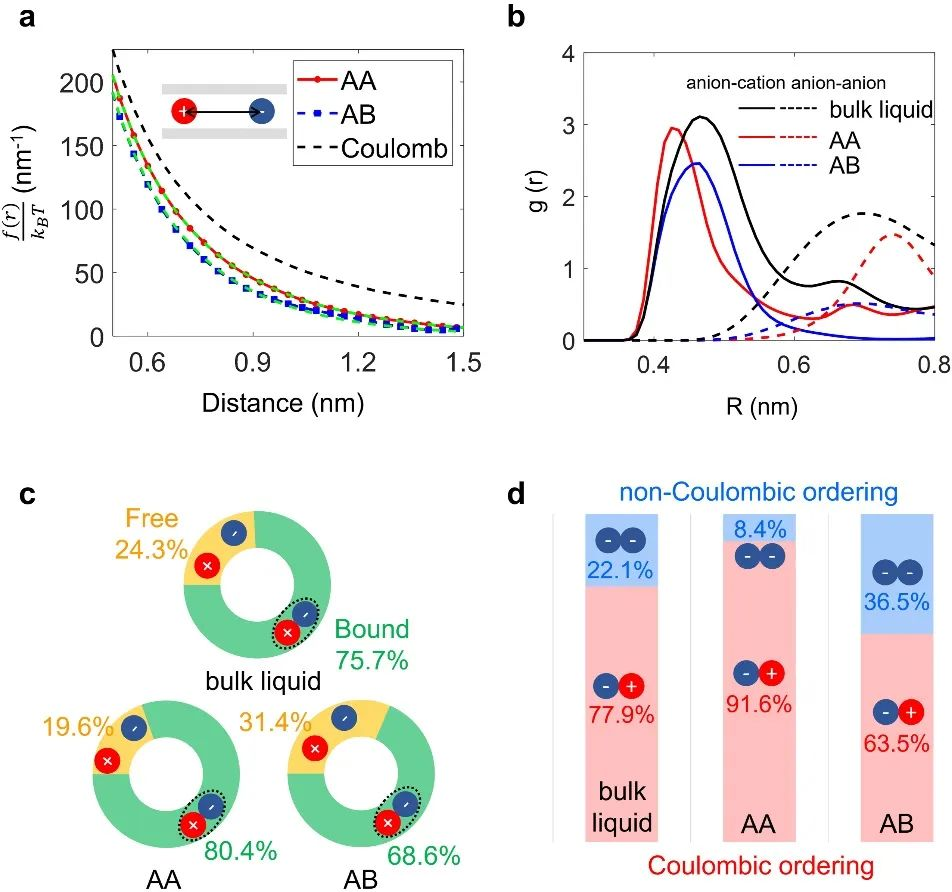
DOI:10.1002/adma.202301118
总结
离子液体是由阳、阴离子构成的液态盐,因缺乏传统溶剂的屏蔽而表现出强烈的离子–离子相关与多体相互作用,导致其局域结构、动力学与宏观输运高度耦合。
要理解并设计这种复杂体系,必须依靠多尺度计算:以DFT提供电子结构与局域相互作用的基准,经典MD刻画热力学、扩散与界面分层,QM/MM还原涉及键重排或电荷转移的化学过程,粗粒化模型揭示长时空下的自组装与输运通道,机器学习则加速性质预测与候选分子筛选。
将这些工具与严格的不确定性评估及实验闭环结合,能把从分子机理到定向分子设计的理念转化为可行的工程应用。