

金属有机框架(Metal-Organic Framework,简称 MOF)是一类新型的多孔材料,它是由金属离子或金属簇与有机配体通过配位键连接而成的晶体结构。可以把它形象地理解为一种“搭积木”的过程:金属节点好比积木的连接点,有机配体则像是连接这些节点的“桥梁”,通过规则的连接方式,它们可以自组装形成各种各样的三维网络结构。
下图展示了常见的MOF结构及其衍生物;
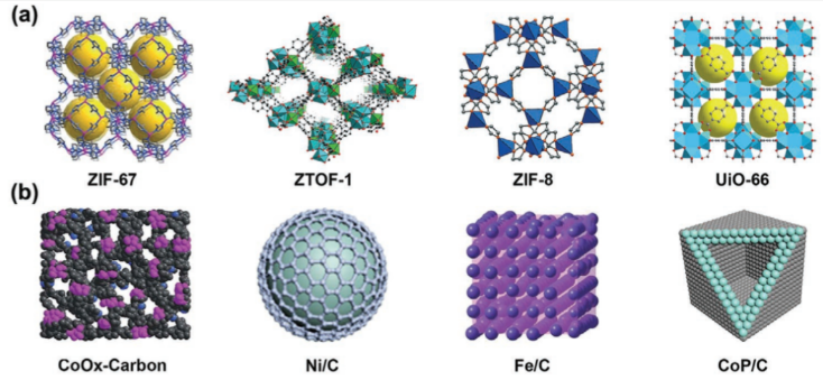

图1 常见的MOF结构及其衍生物。DOI: 10.1002/adfm.202100470
与传统的多孔材料(如活性炭、沸石)相比,MOF拥有极高的比表面积、可调控的孔径以及丰富的化学功能位点。这意味着我们不仅能控制它的孔道大小,还能通过改变金属中心或有机配体来赋予它不同的化学性质。
从宏观角度看,MOF就像是一种高科技“海绵”,能吸附、存储、分离或传输各种分子。
但从微观结构来看,它是一种精密的分子建筑。计算化学在这里扮演着关键角色,因为 MOF的性能与其结构密切相关,而通过实验逐个合成并测试其性能往往费时费力。
借助计算化学方法,我们可以提前预测其结构、稳定性和功能,从而大大缩短材料设计的周期。这也正是MOF材料近年来得以快速发展的重要原因之一。
MOF材料近年来之所以受到科研界和工业界的广泛关注,是因为它们在许多应用领域表现出极大的潜力。
首先,MOF拥有极高的比表面积,有些材料的比表面积甚至可以达到6000 m2/g,这意味着同样质量的材料能提供比普通多孔材料多得多的表面空间,从而吸附更多的气体或分子。
其次,MOF的结构高度可设计化。通过选择不同的金属中心(如Zn、Cu、Fe、Zr 等)和有机配体,我们可以“定制”出具有特定化学功能的MOF,用于特定场景,下图展示了MOFs的合成以及在各个领域的应用,如二氧化碳捕集、氢气储存、药物释放、催化反应等。
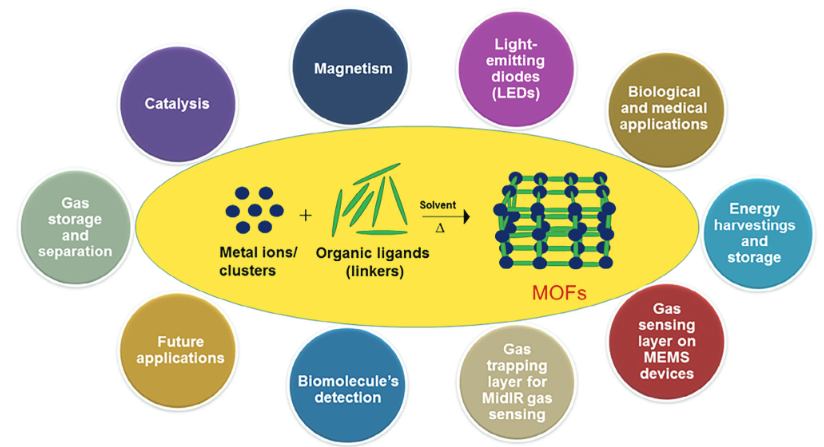
图2 MOF材料的应用场景。DOI: 10.1002/gch2.202300244
从计算化学的角度来看,MOF的受欢迎程度还体现在它是一个“高度可预测”的系统。由于其晶体结构明确、周期性强,适合应用密度泛函理论(DFT)、分子动力学(MD)、蒙特卡罗模拟(MC)等方法来研究其性质。
例如,通过DFT计算,我们能够预测MOF的稳定性、电子结构以及与气体分子的相互作用;通过MD模拟,可以研究分子在MOF孔道内的扩散行为;而通过大规模的高通量计算,我们甚至能够筛选出具有潜在应用价值的候选MOF材料,而无需先行合成它们。
要理解MOF的设计原理,我们可以用“分子积木”的思路来类比。每一种MOF都由两个基本组成部分:金属节点和有机配体。
金属节点通常是金属离子(如 Cu²⁺、Zn²⁺、Zr⁴⁺)或金属簇,它们提供了配位位点;
有机配体则往往是含有羧基、吡啶、咪唑等官能团的有机分子,用于与金属中心形成配位键。
当这两者在特定条件下(如溶剂、温度、pH)混合时,会通过自组装过程生成规则的晶体结构。
在实验室中,MOF的合成方法主要有溶剂热法、水热法、溶剂挥发法等。这些方法各有优势,可以根据具体需求选择合适的方法进行MOF晶体的合成。而在计算化学领域,则可以通过自动数据挖掘和机器学习实现MOF合成预测。
下图展示了MOF逆合成设计(从晶体结构到合成条件)的完整ML工作流程,
1)从关于MOF合成条件及其结构信息的科学文献的自动数据挖掘开始,
2)建立和训练ML模型,
3)预测新MOF结构的合成条件并与人类专家的预测进行比较。
我们的方法标志着从基于经验和启发式的试错方法过渡到MOF合成中的逆合成设计方法的起点,最终在自动化实验室中实现完全自主的MOF发现。
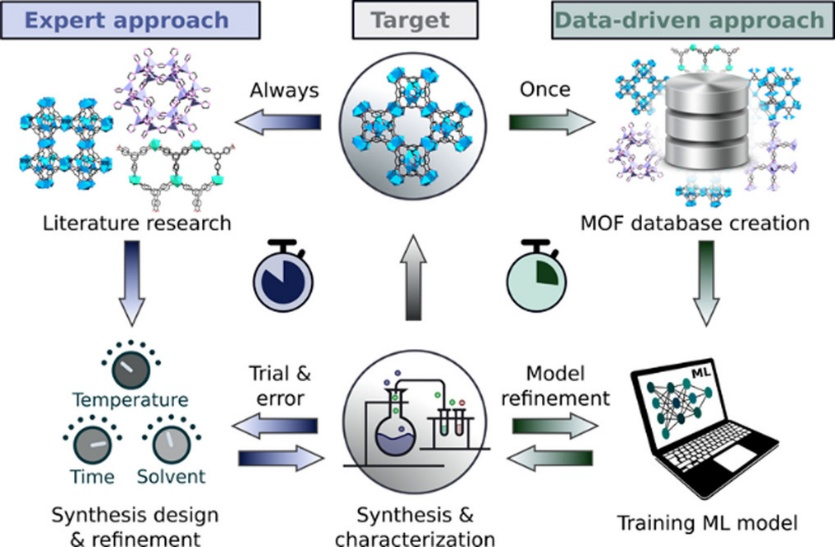
图3 MOF材料逆合成设计的完整机器学习工作流程。DOI: 10.1002/anie.202200242
计算化学在MOF研究中的作用,可以分为几个层次。
第一层是结构预测与优化。通过量子化学计算(尤其是密度泛函理论),我们可以预测不同金属中心和配体组合下的结构参数,如晶格常数、键长、键角等;这有助于研究者筛选出稳定的结构候选。例如下图展示了FJI-H14 MOF通过DFT优化显示三个优先CO2吸附位点。
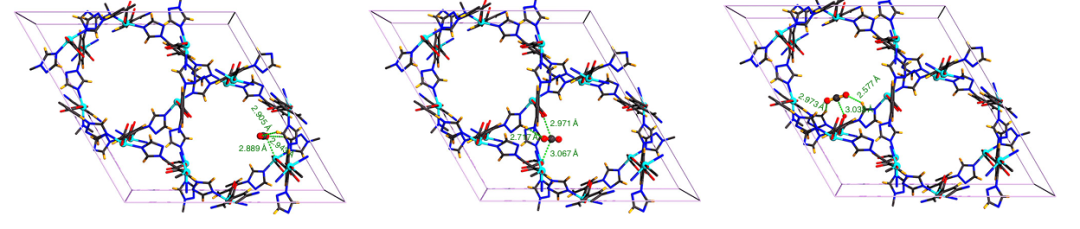
图4 DFT优化显示的MOF材料的三个CO2吸附位点。DOI: 10.1038/s41467-017-01166-3
第二层是性质预测。MOF的吸附性能、催化活性、离子传导性等往往与其孔道结构、电荷分布和表面化学有关,而这些性质可以通过计算模拟获得。
例如,利用蒙特卡罗方法可以计算气体吸附等温线;利用分子动力学方法可以分析分子在孔道内的运动轨迹和扩散系数;利用电子结构计算可以研究其导电性或光催化性能。巨正则系统蒙特卡罗模拟(GCMC)通过随机粒子插入/删除/移动模拟恒化学势系统。下图展示了通过GCMC与DFT揭示Zn基MOF(GUPT-1)中CO2与-NH2基团的静电作用(O-N距离2.925–2.954 Å)。
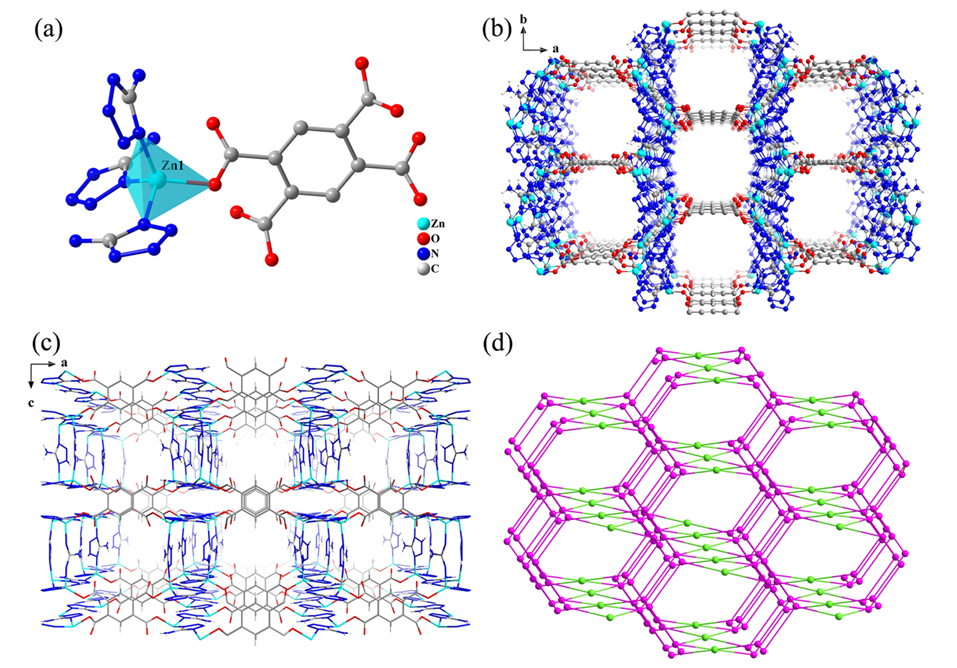
图5 GCMC与DFT协同揭示MOF中CO2与-NH2基团的静电作用。DOI: 10.1021/acs.organomet.2c00054.
第三层是高通量筛选与机器学习。在MOF研究中,已有成千上万种已报道的结构数据库,如CoRE MOF数据库、CSD MOF子库等。通过高通量计算,我们可以在这些数据库中快速评估不同材料的性能;再结合机器学习算法,还能从已有数据中“学习”出结构—性能关系,进而预测尚未合成的MOF的潜在性能。
这种方法近年来已广泛用于气体分离、储氢、CO2捕集、甚至电化学储能领域,使得MOF设计逐渐走向数据驱动和智能化。下图展示了从CoRE MOF数据库中使用ML辅助发现性能最好的C3H8选择性MOF的工作流程示意图。
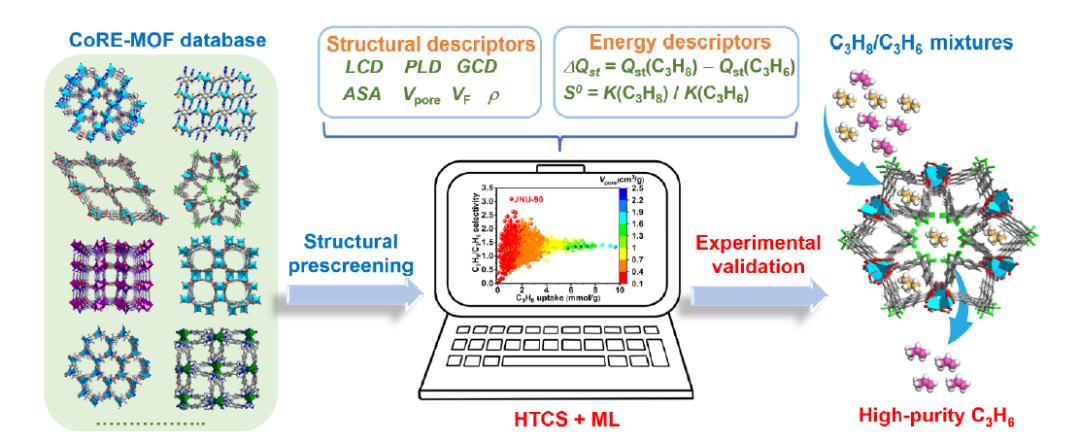
图6 使用机器学习辅助发现性能最好的MOF流程图。DOI: 10.1021/jacs.3c14610
尽管MOF已经在实验室和计算领域取得了巨大进展,但它仍面临一些挑战。
首先是稳定性问题:部分MOF在高温、潮湿或酸碱环境中容易分解,这限制了它们在工业环境中的长期应用。
其次是规模化生产与成本:虽然实验室合成方法灵活,但要实现大规模制备仍存在技术与经济障碍。
此外,从计算化学的角度来看,MOF的研究也面临着计算复杂度的问题。随着 MOF结构越来越复杂,单一的量子化学计算可能无法应对大体系,需要多尺度模拟方法的介入,例如将量子力学计算与经典分子动力学结合,或利用粗粒化模型来研究宏观行为。
未来,MOF的研究很可能会与能源、环境和生命科学等领域深度结合。例如,在电化学储能中,MOF可作为电极材料、离子筛选膜或前驱体材料;在环境治理中,可用于高效捕获温室气体;在生物医药中,可作为智能药物释放载体。
而计算化学将在这一过程中继续发挥不可替代的作用——它不仅能帮助我们理解“为什么这种MOF表现这么好”,还可以预测“下一个更好的MOF应该是什么样子”。随着计算资源的提升、算法的发展和实验验证的反馈,MOF材料的设计将会越来越像“量身定制”,从而加速这一领域从实验室走向实际应用的步伐。
金属有机框架(MOF)是一类由金属节点与有机配体通过配位作用自组装形成的高度多孔晶体材料,因其结构可设计、比表面积极高和功能位点可调控而在气体吸附、储能、催化以及药物释放等领域展现出巨大潜力。
本文从计算化学角度介绍了MOF的概念、性能优势、构建原理以及研究方法。计算化学在MOF研究中发挥着不可替代的作用,它不仅能够通过密度泛函理论(DFT)、分子动力学(MD)和蒙特卡罗(MC)模拟等方法预测材料的结构稳定性和性能,还能通过高通量计算和机器学习实现材料的快速筛选与智能化设计,从而显著缩短实验周期并提高研究效率。
未来,MOF的发展将更多地与能源、环境和生命科学结合,但其在稳定性、规模化生产和计算复杂度方面仍面临挑战,需要多尺度模拟和跨学科策略的进一步支持。随着计算技术和实验验证的协同进步,MOF材料有望实现从基础研究到实际应用的快速转化。