

说明:本文华算科技系统概述DFT的数学基础、泛函分类框架、优缺点与选择策略、跨学科应用及前沿发展,为后续分析奠定理论基础。


密度泛函理论的基础概念与原理
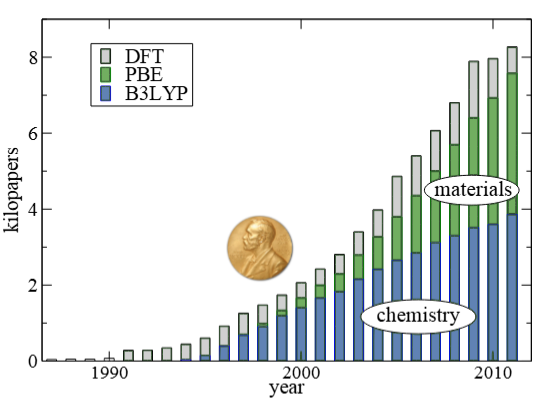
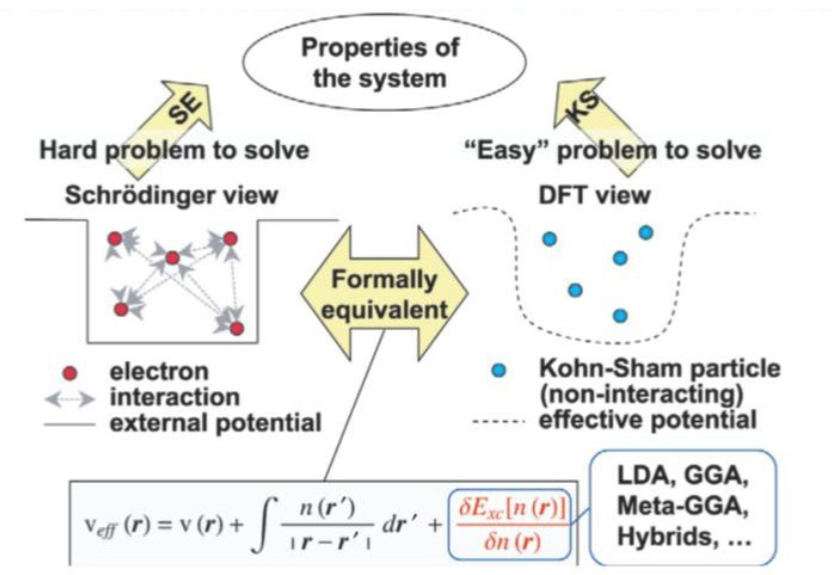
密度泛函理论(Density Functional Theory,DFT)是计算化学和材料科学中最为广泛应用的第一性原理计算方法,它通过电子密度来描述多电子系统的基态性质,而非传统的波函数方法。
DFT的核心思想在于:系统的所有性质都可以由电子密度唯一确定,这大大简化了量子力学计算的计算复杂度。与基于波函数的传统方法(如Hartree-Fock)相比,DFT在计算效率和准确性之间取得了良好平衡,使其能够处理包含数百个原子的复杂体系。
DOI: 10.1063/1.4704546
常见泛函的类型、特点与优缺点分析 局域密度近似(LDA)
常见的LDA泛函包括Xα(Slater交换)、VWN(Vosko-Wilk-Nusair)和PW(Perdew-Wang)等。其中VWN3和VWN5是应用最广泛的LDA相关泛函,而PW泛函在计算石墨层间弱相互作用时表现优于其他LDA泛函。
优点:计算效率极高,适用于金属体相性质预测(如晶格常数误差);对均匀电子气严格成立。
缺点:系统性低估键解离能和高估结合能、对分子体系过度离域化,导致能隙严重低估(如半导体Ge的带隙预测值仅为实验值50%)、无法描述范德华力等非局域效应
广义梯度近似(GGA)系列
GGA泛函可分为两大类:交换泛函(如B88、PW91、PBE)和相关泛函(如LYP、P86、PBE)。其中PBE(Perdew-Burke-Ernzerhof)泛函是最著名的无参数GGA泛函,所有参数都由物理条件确定,具有很好的转移性能。
GGA泛函相比LDA的主要改进包括:更准确的反应能垒预测、改进的分子几何结构和振动频率、对氢键等弱相互作用的更好描述。然而,GGA泛函仍然会严重低估电荷转移激发、里德堡激发等需要正确渐进行为的体系,也明显低估带隙。
杂化泛函与meta-GGA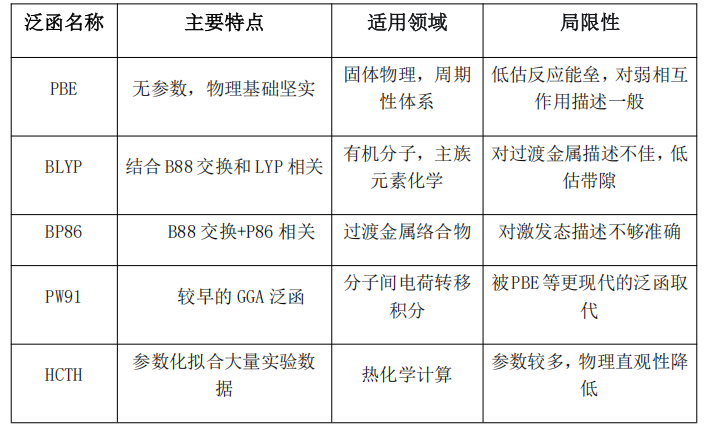
meta-GGA泛函(如TPSS、M06-L、SCAN)进一步引入了动能密度,提供了更高的灵活性和准确性。这些泛函通常包含更多的物理约束条件,能够在不同体系中保持较好的准确性。
近年来,范围分离杂化泛函(如CAM-B3LYP、ωB97XD)成为研究热点,它们通过将电子–电子相互作用分为短程和长程两部分,分别用DFT和HF方法处理,解决了传统杂化泛函在描述电荷转移激发等方面的困难。
顶刊案例研究:DFT在电催化中的应用 研究背景与意义
2021年,昆明理工大学王华教授团队在Chemical Society Reviews上发表了题为“Density Functional Theory Studies of Transition Metal Carbides and Nitrides as Electrocatalysts”的综述论文,系统总结了DFT在过渡金属碳化物和氮化物电催化剂研究中的应用。
这项工作之所以能发表在顶级期刊,因为它首次从原子–分子层面系统分析了过渡金属碳化物及氮化物基催化剂在六个经典电化学反应(析氢反应HER、氧氧化反应OER、氧还原反应ORR、氮还原反应N2RR、CO2还原反应CO2RR和醇氧化反应)中的结构–性能关系。

DOI: 10.1039/d1cs00590a
计算方法与创新点
该研究采用第一性原理计算方法,基于密度泛函理论(DFT)框架,使用VASP(Vienna Ab initio Simulation Package)软件包进行电子结构计算。
计算采用了PBE泛函(属于GGA类),并考虑了DFT-D3方法进行色散校正。对于强关联体系,采用了DFT+U方法修正电子相关效应。
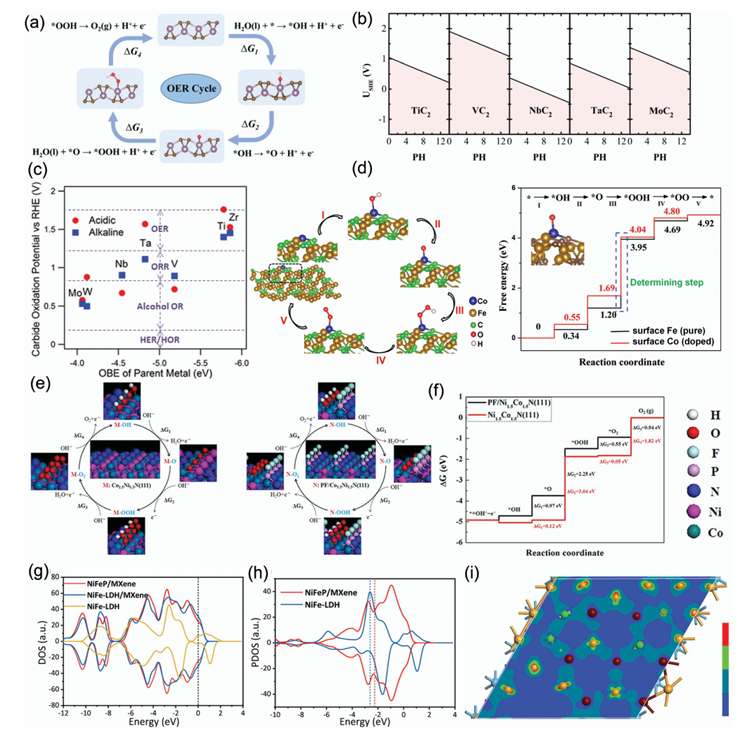
研究者特别关注了描述因子(Descriptors)的建立,这是连接催化剂微观电子结构与宏观催化性能的关键桥梁。
例如,对于析氢反应(HER),催化剂的活性与氢吸附自由能(ΔG)存在Volcano型关系;对于氧还原反应(ORR)和氧析出反应(OER),活性与氧中间物种(O、OH、OOH)的吸附能密切相关
DOI: 10.1039/d1cs00590a
该研究通过系统的DFT计算,揭示了过渡金属碳化物和氮化物的催化活性起源。例如,研究发现镍氧化物(NiO)的{1,1,1}晶面和高价Ni3+可以优化H2O2选择性,而钴单原子催化剂(Co-N2O2)通过环氧氧调控*OOH吸附强度,实现高效2e-ORR路径。
总结
本文系统剖析了LDA、GGA、杂化泛函的物理基础、性能边界与适用场景,结合过渡金属催化、光材料设计等典型案例揭示泛函选择的核心逻辑。
通过融合最新进展(如SCAN、机器学习泛函),读者将具备解决材料设计、催化机制等前沿问题的实战能力,规避常见计算陷阱,显著提升研究效率与准确性。