

说明:本文华算科技将深入剖析化学世界的三大核心作用力——离子键、共价键、金属键,不仅厘清其基本概念,更将探讨其现代计算模拟方法、实际应用场景,并结合顶级科研期刊的最新案例,为您展现一个既基础又前沿的化学键世界。
在我们周围,从坚不可摧的金刚石到柔软可塑的钠金属,从晶莹剔透的食盐到生命之源的水分子,万物皆由原子构成,而将这些原子“粘合”在一起、塑造大千世界无穷性质的,正是化学键。理解化学键,就如同拿到了开启材料科学、药物设计、纳米技术等领域的钥匙。
不同类型的化学键定义
离子键:通过静电吸引力结合带相反电荷的离子(如阳离子与阴离子)。金属元素倾向于失去电子形成正离子,非金属元素获得电子形成负离子,通过库仑力结合成晶体结构。(如Na+与Cl–静电吸引)
共价键:原子间通过共享电子对形成,电子云重叠使原子稳定结合。常见于电负性相近的非金属元素(如H2O中的O-H键)。
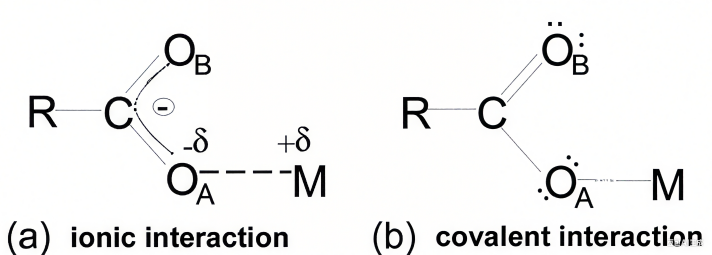

图1 离子相互作用与共价相互作用的区别 DOI: 10.1002/ange.201902229
金属键:金属原子释放价电子形成“电子海“,带正电的金属离子沉浸在自由电子云中,通过电子–离子间静电吸引结合。
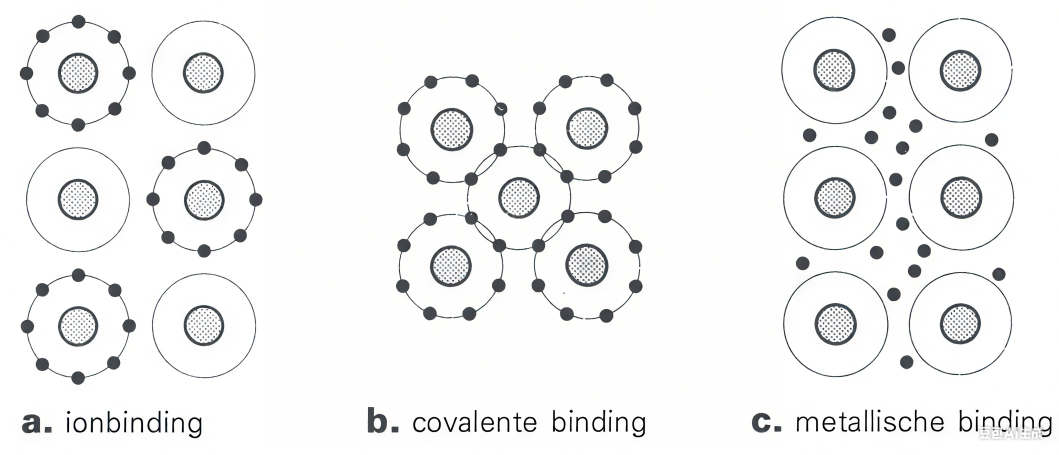
图2 a.离子键模型 b.共价键模型c.金属键模型
化学键的计算
离子键
经典计算(晶格能):它可以通过玻恩–朗德公式或卡普斯钦斯基方程进行估算,这些公式综合考虑了离子电荷、离子半径、晶体构型等因素。
量子化学计算:现代计算化学软件(如VASP, Gaussian)使用密度泛函理论(DFT) 等方法,通过求解薛定谔方程来精确计算离子晶体的电子结构、结合能、电荷分布等,从而深入揭示离子键的本质。例如,可以通过Bader电荷分析来量化电子转移的程度,精确判断每个离子所带的电荷量。
共价键
价键理论(VB):由海特勒和伦敦解H2分子薛定谔方程开创,鲍林发展。该理论强调电子配对和原子轨道杂化。它直观地解释了键的形成、键角和分子的空间构型(如CH4的正四面体结构源于sp3杂化)。
分子轨道理论(MO):更现代和强大的理论。它将所有原子轨道线性组合成属于整个分子的分子轨道。电子不再定域于两个原子之间,而是在整个分子中离域分布。该理论成功解释了O2的顺磁性等价键理论无法解释的现象。药物设计:过渡态类似物
DFT计算:DFT计算是研究复杂共价体系(如蛋白质、催化剂)的黄金标准。它可以可视化分子轨道、计算键级、键能、以及进行电子密度拓扑分析(AIM理论),精确描绘共价键的临界点和路径。
金属键
自由电子模型:最简单的模型,将价电子视为在势阱中自由运动的粒子,成功解释了金属的导电和导热性。
能带理论:量子力学处理金属键的框架,是分子轨道理论在晶体上的扩展。原子轨道组合形成能量连续分布的能带。价带(被电子填满)和导带(空或部分填充)之间的重叠关系决定了材料的导电性:金属的导带是部分填充的,电子可以轻易跃迁和移动。
DFT计算:可以精确计算金属及其合金的能带结构、态密度(DOS)、费米能级以及结合能,用于预测新金属材料的稳定性、力学性能和电子性能。
可视化分析化学键
静电势
计算分子表面静电势具有重要理论意义,该性质可用于准确识别分子中的亲电性与亲核性区域,对深入理解离子键、氢键、配位键等化学作用中的静电贡献至关重要。
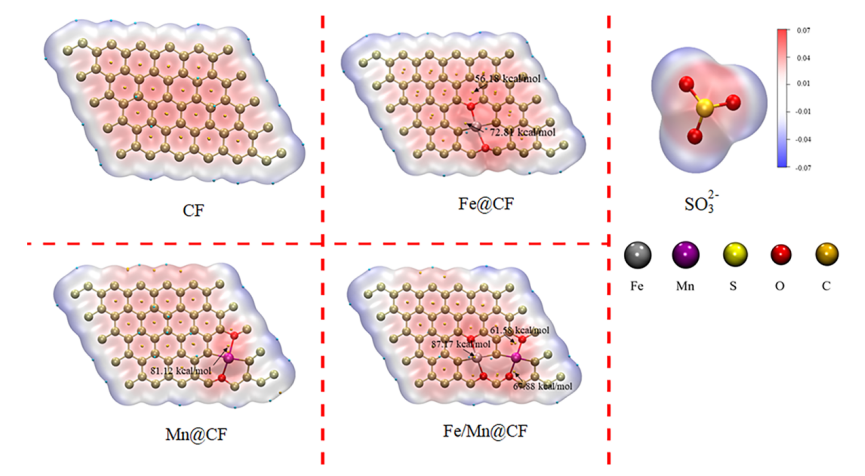
DOI:10.1016/j.cej.2024.152342
在化学反应过程中,分子表面静电势的分布特征能够显著调控分子间相互作用模式,例如决定分子间呈现吸引或排斥行为,进而影响反应路径的选择性,是决定反应可行性及具体反应路径的关键因素之一。
IGMH分析
IGMH(基于Hirshfeld分割的独立梯度模型)是一种用于化学体系成键与相互作用分析的可视化方法,其核心优势在于能够清晰表征弱相互作用(如氢键、π–π堆积和范德华力等)的空间分布与强度,并通过等值面图谱与着色方案直观展示作用区域。
该方法也可用于化学键性质的判别,有效区分共价键、离子键等不同键型并评估其强度。此外,IGMH适用于周期性体系(如晶体结构与表面吸附过程),能够充分考虑周期边界条件,从而准确分析层间相互作用或吸附行为。
IGMH方法将可视化表达与定量分析相结合,在复杂化学体系及弱相互作用研究方面表现出显著优势,成为一种强有力的理论化学工具。
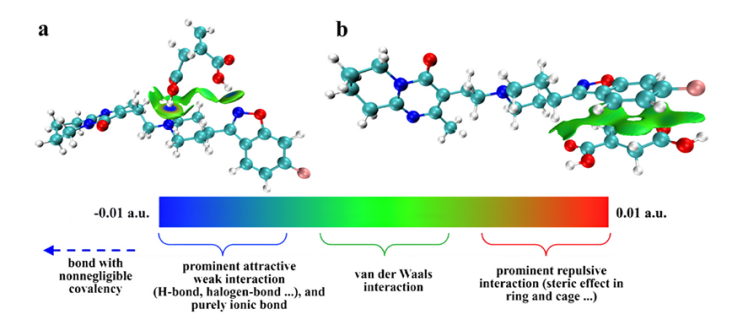
DOI:10.1016/j.envres.2025.121296
分子轨道分析
通过分子轨道理论计算,能够以量子化学方法精确绘制分子轨道的空间分布图像,直观展示成键轨道、反键轨道与非键轨道的电子云密度分布特征,进而揭示分子中电子的离域行为与键合性质。
该可视化方法对理解共轭体系中的π键形成、芳香性判据以及分子反应活性等关键概念具有重要理论价值。
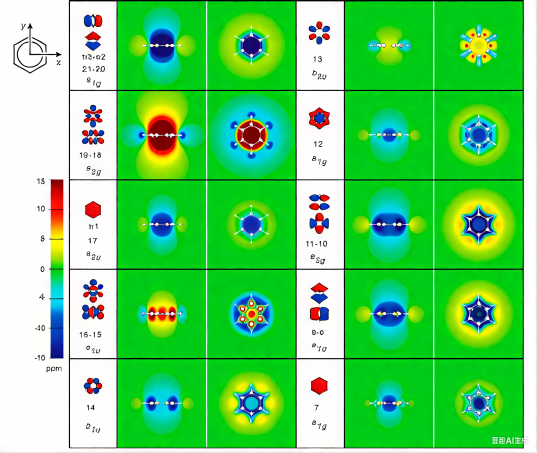
DOI:10.1021/jp411410r
以苯分子为例,其分子轨道的三维等值面图可清晰呈现出高度离域的π分子轨道系统,显示具有D6h 对称性的环状电子云分布与完全离域的π电子共轭体系,从而在量子力学层面为苯的芳香性——如符合Hückel规则、具有显著共振能及磁各向异性效应,提供了直接的图形化证据。
案例分析
用于低温应用的抗断裂高熵合金
研究简介:传统金属材料在极低温下通常会变脆。这项研究报道了一种CrMnFeCoNi高熵合金(HEA),它在液氮温度(77 K)甚至液氦温度(20 K)下,不仅强度增高,韧性和抗断裂性也显著增强,这与几乎所有已知材料的行为相悖。
图3. 在低温下,高熵合金的变形机制从位错滑移转变为产生大量纳米孪晶(TWIN),有效阻碍裂纹扩展,表现出超高韧性)
DOI: 10.1126/science.1254581
这种非凡性能的根源在于其多主元特性引起的复杂金属键环境。多种不同大小的原子高度无序地分布在晶格中,产生了严重的晶格畸变。这种畸变极大地阻碍了位错(晶体中的缺陷线)的运动,从而提供了异常强化的基础(固溶强化)。
更重要的是,在低温变形时,其塑性变形机制会从位错滑移诱导转变为纳米尺度的机械孪生,这种孪生能有效地钝化裂纹尖端,吸收能量,从而在原子尺度上阻止裂纹扩展。这一切都源于其独特的、由多种元素共同贡献的金属键网络所提供的多重能量耗散路径。
总结
化学键作为化学学科的核心理论框架,构成了原子通过相互作用形成分子与凝聚态物质的桥梁。
随着理论计算方法的发展,尤其是基于量子力学的电子结构理论的广泛应用,我们得以从电子结构层面深入揭示化学键的本质,并实现对键型、键强及键性质的精确定量描述与可视化分析。
借助第一性原理计算、电子密度拓扑分析(如AIM理论)、以及反应性描述符(如分子静电势)等多种理论工具,可以系统解析化学键的成键机制、稳定性和电子分布特征,极大地深化了对化学键多维度物理图像的理解。