

Q1:朱老师,这种扩散能垒的,它的能垒是1.214还是1.756啊?
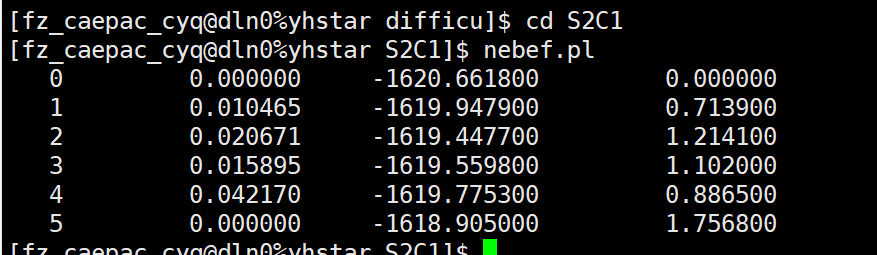

A:没有能垒,中间能量都没有末尾高
Q2:朱老师,一个任务可以先放任它跑 100 步,没有收敛的迹象的话,停下来,得到电荷密度和波 函数后重新计算。 这里得到的电荷密度和波函数能当结果直接用吗?
A:后续收敛的可以,没收敛不行
Q3:朱老师,请问为什么我用split_dos分解出来的总态密度dos0文件只有四列数据,而且作图出来不太对,其他dos1什么的都正常
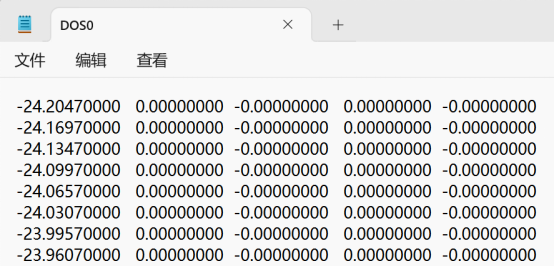
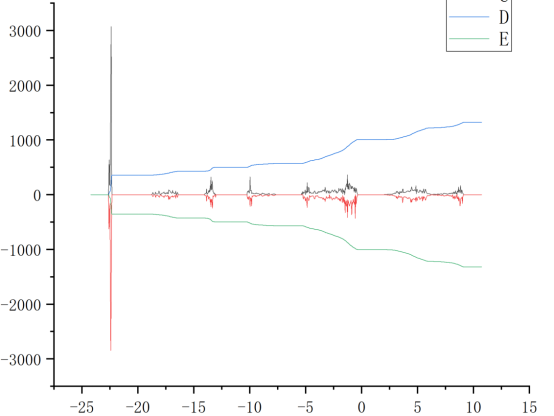
A:没有问题的,123列作图,调整一下横纵坐标的显示
Q4:朱老师,我问一下这个过渡态是指的什么?这个过渡态跟最后一步的图不都是差不多的吗,有什么区别呢?
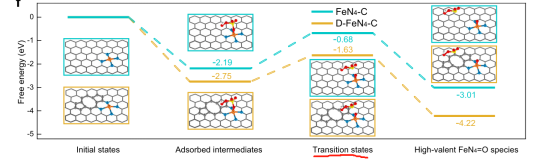
A:过渡态是化学键将断未断时的状态,区别在于末态分子离单原子更远了
Q5:朱老师,正极材料脱锂后的结构优化计算,要放开晶格优化吗(ISIF=3)?
A:晶胞大的话用2,不算大的话用3
Q6:朱老师,这是我计算的六系三元材料的总DOS图,是将态密度分开后的dos0文件,为什么在费米能级处无带隙,而在1eV——5eV附近出现一个较大的带隙,这是合理的吗?
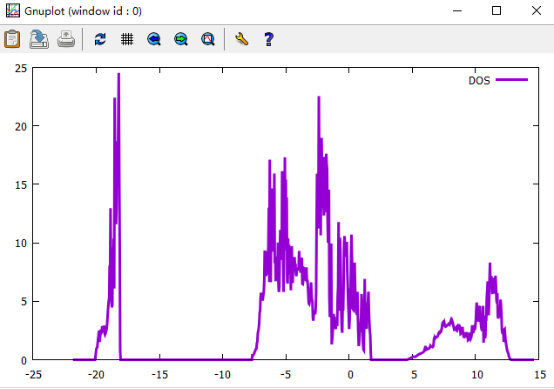
A:这是金属性的,要看费米能级对应的态密度强度
Q7:朱老师,您帮我看看我这个是怎么回事,为什么没有出现 charge spilling?
A:计算仍在进行,还没有算完
Q8:朱老师这个总态密度的0点为啥咋直线的右方,问题可能出现在什么地方?
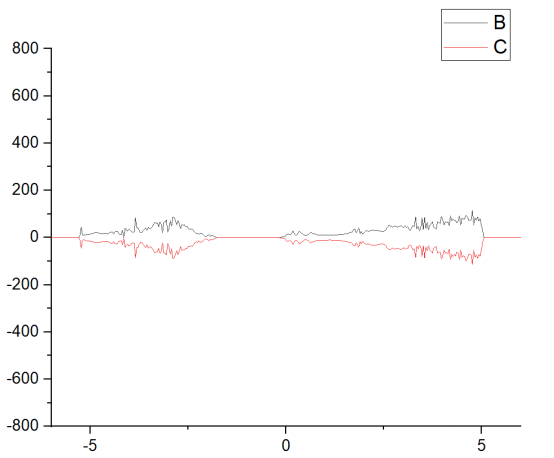
A:费米能级不一样,是不是有缺陷之类的,N缺陷就会上移,P缺陷下移
Q9:朱老师,VASP计算得到的CHGCAR导入VESTA绘制电荷密度图时,isosurface value的单位是什么呢?
A:E/bohr^3
Q10:朱老师,审稿人要求我加一个计算,计算水解离的第二步反应(Tafel路径*H + *H → H2+* 和Heyrovsky路径*H + H2O+ e- → H2 + OH- + *),但是我在计算这两个过程的过渡态的时候结果一直不收敛,现在快到了交稿时间,可不可以不计算过渡态,直接给出这两步反应的能垒,我想知道这样的计算过程、结果合不合理?
A:不收敛也没办法了,只能给自由能不给过渡态了