目的: 说明如何使用Adsorption Locator模块计算结合能以评估添加剂的应用潜力。 所用模块: Materials Visualizer, Adsorption Locator, Forcite, COMPASS 先决条件: 掌握可视化晶体模型建立Crystal Builder Visualizer教程 晶体块体的几何外形对许多工业过程至关重要。在化学和制药工业中的以下过程中,晶体形状均是重要因素,体现在以下方面:
因此,晶体形态与晶体内部原子排列之间的关系是化学家、化学工程师和工艺工程师非常感兴趣的问题,由此可以预测晶体的形状,开发特制的添加剂,控制溶剂和杂质的影响等。因此,添加剂对晶体生长和晶体形貌的影响是非常重要的。Adsorption Locator模块可以模拟添加剂在晶面上的吸附,从而研究吸附过程的能量变化机理及其对晶体生长的影响。
介绍
颜料红(1,4-二酮吡咯(3,4-c)吡咯的二苯基衍生物,DPP)是一种优质杂环颜料,具有良好的热稳定性、着色强度和遮盖力,以及优异的耐光性和耐候性。通过在生产过程中控制颗粒大小,可以制备出透明或不透明的晶体。
在本教程中,将研究表面上官能团之间的距离。结果表明,一种氨基酸衍生物可以作为生长抑制剂
本教程包括如下部分:
注意:为了确保您可以完全按照预期的方式学习本教程,您应该使用“设置管理器(Settings Organizer)”对话框确保项目中所有参数都设置为BIOVIA的默认值。
1、开始
首先启动Materials Studio并创建一个新项目。 打开“新建项目New Project ”对话框,输入Pigment Red Additive 作为项目名称,单击“确定OK ”按钮。
Pigment Red Additive 在项目浏览区域Project Explorer中列出。现在,导入要研究的输入文件。
在Project Explorer中,右键单击项目名根目录并选择Import… 打开“导入文档Import Document”对话框。导航到Examples/Documents/3D Model 文件夹,选择pigment_red_010.xsd 文件,然后单击Open 按钮。
在该部分中,将从(0 1 0)颜料红表面构建一个平板模型(具有真空层的表面)。晶体模型是通过利用在表面层之间,引入大于表面层厚度的真空区域,在给定方向上重复表面结构来构造的。表面必须足够大,可容纳所研究的特定抑制剂。 从菜单栏中选择Build | Symmetry | Supercell ,以打开Supercell 对话框。将U 和V 分别增加到4 和6 ,单击Create Supercell 按钮并关闭 对话框。
这将构建出扩大的表面结构,现在可以从二维表面模型构造三维平板模型。
从菜单栏选择Build | Crystals | Build Vacuum Slab… ,打开“构建真空层Build Vacuum Slab Crystal”对话框。在Vacuum Slab 选项卡上,将“真空层厚度Vacuum thickness ”更改为50 Å,然后单击Build 按钮。 单击3D Viewer工具栏上的“显示样式Display Style ”按钮 以打开Display Style对话框。在Lattice 选项卡上,将C的最大值(Range Max. C )设置为2.00 。
可以预览到真空层与表面层在c轴方向上的排列。
在本教程的后面部分,将在表面上添加添加剂。由于周期性边界条件,添加剂可能与两个表面发生相互作用。因此,需要在表面上方添加一个较大的真空区域,以便添加剂只能与其中一个表面发生相互作用。在本例中,(0 1 0)表面的厚度约为20 Å,因此50 Å的真空层厚度就足够了。
将Range Max. C 更改回1.00 ,然后关闭 Display Style对话框。从菜单栏中选择File | Save Project 保存项目中的所有文件,然后选择Window |Close All ,关闭已经打开的窗口。
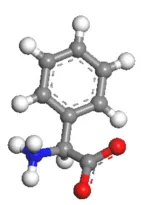
本教程中使用的抑制剂2-苯基甘氨酸是一种以两性离子形式存在的氨基酸衍生物。
单击New 按钮后的箭头 并从下拉列表中选择3D原子文档3D Atomistic Document 。在Project Explorer中,右键单击3D Atomistic.xsd ,然后从快捷菜单中选择“重命名Rename ”。将文档的名称更改为inhibitor.xsd 。使用草图绘制Sketch 工具栏上的工具构建2-苯基甘氨酸分子(如下所示),然后使用Clean 工具 释放应力,使得结构更加合理。 抑制剂2-苯基甘氨酸的分子结构
现在,使用Forcite模块和COMPASS力场优化抑制剂的分子构型。
从菜单栏中选择Modules | Forcite | Calculation 以打开Forcite Calculation对话框。从“任务task ”下拉列表中选择“几何优化Geometry Optimization ”。在Energy 选项卡中,设置“力场Forcefield ”为COMPASSIII ,对于静电Electrostatic和范德华相互作用van der Waals的求和方法Summation methods ,均选择基于原子截断(Atom Based )。在“作业控制Job Control ”选项卡中,把“网关Gateway location ”选择为“我的电脑My Computer ”并点击“运行Run ”。 这将在Project Explorer中创建一个名为inhibitor Forcite GeomOpt的新文件夹。计算过程将仅花费不到一分钟的时间。计算完成后,能量最小的构型将被保存在文件夹中的inhibitor.xsd文件,并显示在Materials Visualizer中。 从菜单栏中选择File | Save Project 并关闭所有打开的文档。
在上一个步骤中,已经构建了颜料红(0 1 0)表面的3D平板模型,并优化了抑制剂的结构,现在将抑制剂分子结合在表面结构上。
在Project Explorer中,双击pigment_red_010.xsd 文件。 接下来,进行吸附计算,以搜索表面上可能的吸附位点和抑制剂分子的取向。然后对晶体表面的抑制剂分子进行几何优化,以获得全局最小能量的构型。 从菜单栏中选择Modules | Adsorption Locator | Calculation 打开Adsorption Locator Calculation对话框。 在Setup 选项卡中,从计算任务task 的下拉列表中选择模拟退火Simulated annealing 。吸附质Adsorbate 中选择优化过的inhibitor.xsd 结构,计算精度Quality 设置为粗糙Coarse 。 在Energy 选项卡中,选择力场为COMPASSIII ,静电和范德华相互作用的求和方法Summation methods 均选择基于原子截断(Atom Based )。 注意:对于非键项能量的计算,Ewald求和方法精确度更高,但需要大量的CPU计算时间。在本教程中,使用基于原子截断的求和方法和较低的计算精度以加速计算。 可以通过多种方式限制搜索低能量构型的空间。在本教程中,将该空间定义为在表面上氢键施主和受主10 Å的范围内。 在Location 选项卡中,勾选“由原子集合定义的表面区域(Surface region defined by atom set )”复选框。 将表面上的氢键部分定义为抑制剂可能发生吸附的位置。 在pigment_red_010.xsd 中,旋转并放大其中一个晶体层的表面。
按住Q+SHIFT 键,点金鼠标左键拖动使用套索工具选中分子顶层的所有酰胺基团amide 。 在Adsorption Locator Calculation 对话框的Location 选项卡中,单击“将选定原子添加到目标原子集合Add selected atoms to TargetAtoms se t”最开始的Add按钮。选中“设置最大吸附距离Set maximum adsorption distance ”复选框,然后输入值10.0 。 单击Run 按钮运行计算并关闭对话框。
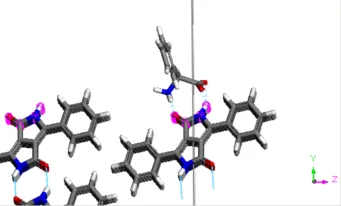
这将在Project Explorer中创建一个名为pigment_red_010 Adsorption Anneal的新文件夹。根据计算机处理器的速度,计算可能需要一些时间才能完成。 计算运行时,包含各种能量贡献的图表将实时更新。文本文件Status.txt显示计算时间和到目前为止完成的计算步数。计算完成后,pigment_red_010.xsd文档将呈现计算好的结果构型。 等待计算任务完成后再进行结果分析处理。计算完成后,将返回一个包含许多低能量构型的数据表,以及每个构型的能量特性。 打开数据表pigment_red_010.std,双击第一行中的结构。 通过显示氢键,可以观察抑制剂和表面酰胺基团之间的相互作用。 单击计算氢键按钮Calculate Hydrogen Bonds 。
附有抑制剂的晶体表面
数据表的第三列吸附能包含与吸附最相关的能量参数,包括两部分: 第二列的总能量Total energy 包含吸附能加上吸附质的内能,不包括晶格的能量。 最后一列F 是差分吸附能,即去除特定组分的吸附质的能量。因为在本例中只有1个分子和1个组分,该列的值与C列相同。 (0 1 0)晶面的附着能已经计算过,约为-27 kcal/mol。如果要使用Materials Studio计算获得该能量,则可利用Morphology模块进行,在“颜料红的形貌预测”教程(Morphology prediction for Pigment Red)中将详细介绍计算步骤。 对于(0 1 0)晶面,由Adsorption Locator计算的吸附能比附着能呈现绝对值更大的负值。因此,2-苯基甘氨酸是抑制颜料红晶体(0 1 0)晶面快速生长的一个很好的候选物,有助于晶面生长更等距的形态。
从菜单栏中选择File | Save Project ,然后选择Window | Close All 。 参考文献:
Hartman, P.; Perdok, W. Acta Crystallogr., 8, 521 (1955).
Bennema, P. J. Cryst. Growth, 166, 17 (1996). 声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!



 以打开Display Style对话框。在Lattice选项卡上,将C的最大值(Range Max. C)设置为2.00。
以打开Display Style对话框。在Lattice选项卡上,将C的最大值(Range Max. C)设置为2.00。